
Abstract
Background
Gastric cancer (GC) is one of the leading causes of cancer deaths with high heterogeneity. There is currently a paucity of clinically applicable molecular classification system to guide precise medicine.
Methods
A total of 70 Chinese patients with GC were included in this study and whole-exome sequencing was performed. Unsupervised clustering was undertaken to identify genomic subgroups, based on mutational signature, copy number variation, neoantigen, clonality, and essential genomic alterations. Subgroups were characterized by clinicopathological factors, molecular features, and prognosis.
Results
We identified 32 significantly mutated genes (SMGs), including TP53, ARID1A, PIK3CA, CDH1, and RHOA. Of these, PREX2, PIEZO1, and FSIP2 have not been previously reported in GC. Using a novel genome-based classification method that integrated multidimensional genomic features, we categorized GC into four subtypes with distinct clinical phenotypes and prognosis. Subtype 1, which was predominantly Lauren intestinal type, harbored recurrent TP53 mutation and ERBB2 amplification, high tumor mutation burden (TMB)/tumor neoantigen burden (TNB), and intratumoral heterogeneity, with a liver metastasis tendency. Subtype 2 tended to occur at an elder age, accompanying with frequent TP53 and SYNE1 mutations, high TMB/TNB, and was associated with poor prognosis. Subtype 3 and subtype 4 included patients with mainly diffuse/mixed type tumors, high frequency of peritoneal metastasis, and genomical stability, whereas subtype 4 was associated with a favorable prognosis.
Conclusions
By integrating multidimensional genomic characteristics, we proposed a novel genomic classification system of GC associated with clinical phenotypes and provided a new insight to facilitate genome-guided risk stratification and disease management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is a common malignancy with high rates of morbidity and mortality worldwide, and nearly two-thirds of GC cases and deaths occur in Asia [1]. At present, China has the largest number of GC patients, in which more than 679,000 new GC patients are diagnosed and about 498,000 GC-related deaths occur annually [2, 3]. GC patients are frequently diagnosed at advanced stages with a low 5-year survival rate. Peritoneal dissemination and liver metastasis are the most important causes of treatment failure [4]. In addition to surgery, chemotherapy, and targeted therapies, immunotherapy has recently been considered as one of the promising treatments for specific subtypes of GC [4, 5]. However, high heterogeneity of GC remains a crucial barrier to optimal treatment strategies for a better survival benefit [4, 6].
Next-generation sequencing (NGS) has dramatically expanded the knowledge of the molecular basis of tumor heterogeneity. Similar to other types of cancer, multiple molecular classification systems of GC have been presented [7,8,9]. Among these studies, the most acknowledged one was The Cancer Genome Atlas (TCGA) classification. By integrating sequencing data from six molecular platforms, TCGA classified GC into four molecular subtypes, including Epstein–Barr virus (EBV), microsatellite instability (MSI), chromosomal instability (CIN), and genomically stable (GS). Notably, genomics was considered as a fundamental basis for simplifying multi-omics data in the TCGA’s study [7]. Other studies also demonstrated the critical role of genomics in precise molecular classification for various types of cancer [10, 11]. Different aspects of in-depth genome-based analyses, such as mutational signature (reflecting the characteristic imprints of mutagenic processes of human cancer) [12] and clonality (i.e., intratumor heterogeneity) [13], have been reported to be clinically relevant in multiple types of cancer. Neoantigens are mutated peptides derived from somatic mutations and represent excellent targets for immunotherapy, due to their specific expression in cancer tissue [14]. Recent advances in genomics and bioinformatics have facilitated identification of neoantigens through cancer genome sequencing [15, 16]. Our previous study preliminarily suggested the correlation between neoantigen load and clinicopathological variables of GC [17]. Therefore, it is promising to integrate these aspects and develop a novel genome-based classification of GC with an improved clinical significance. In addition, patients of TCGA cohort were predominately from Western population, but the epidemiological, histological, and molecular features of GC differ between Asian and Western countries [18,19,20]. Asian GC have higher prevalence of intestinal type tumors, lower prevalence of MSI GC, and more favorable prognosis. Therefore, an Asian-specific genomic classification is necessary.
In the present study, we performed whole-exome sequencing (WES) of 70 GC samples from Chinese population. With an innovative integration of genomic features, such as mutational signature, copy number variation (CNV), neoantigen, clonality, and essential genomic alterations, we divided GC samples into four subtypes according to distinct genomic features, clinicopathological characteristics, patterns of metastasis, and overall survival (OS). Besides, we proposed a new clinically relevant genome-based classification system for GC and provided rationale for genome-guided therapy.
Methods
Patient tissue samples
Primary GC patients who underwent gastrectomy at the First Affiliated Hospital of Zhejiang University School of Medicine between January 2016 and January 2018 were retrospectively procured. Cases were enrolled in this study according to the criteria as follows: at least 18 years old; pathologically confirmed gastric adenocarcinoma; complete clinicopathological and follow-up data; without autoimmune disease or other cancer types; without previous chemotherapy/radiotherapy. The pathologic diagnoses and characteristics were independently determined by at least two experienced pathologists (Mei Kong and Xiaodong Teng). The tumor-node-metastasis (TNM) Staging was according to AJCC 8th edition [21]. The follow-up data were obtained by medical record system, phone, and letter every 3 to 6 months from the date of surgery. A written informed consent was obtained prior to participation for each participant. This study was approved by the ethics committee of the First Affiliated Hospital of Zhejiang University School of Medicine.
DNA extraction and whole-exome sequencing
DNA was extracted from 70 GC tumors and paired normal gastric mucosa. QIAamp DNA Mini Kit (Qiagen) was used to isolate genomic DNA from tumor tissues and matched normal mucosa according to the manufacturer’s instructions. Then we combined the following two steps to verify the quality of isolated genomic DNA. First, DNA degradation and contamination were monitored on 1% agarose gels. Second, Qubit® DNA Assay Kit in Qubit® 2.0 Flurometer (Invitrogen, USA) was used to quantify DNA concentration.
Whole-exome library construction was generated using the Agilent SureSelect Human All Exon V6 Kit (Agilent Technologies, Santa Clara, CA, USA). The index-coded samples were clustered on a cBot Cluster Generation System using Hiseq PE Cluster Kit (Illumina). Then the DNA libraries were sequenced on Illumina Hiseq platform (Illumina, San Diego, California, USA) and 150 bp paired-end reads were generated. We first conducted data quality control and then performed all downstream bioinformatics analyses based on the high-quality clean data, in which reads containing an adapter, reads containing poly-N, and low-quality reads were removed. The paired-end clean reads were aligned to the Human Genome Reference Consortium build 37 (GRCh37) using BWA v.0.7.8 [22]. Mapped reads were then de-duplicated using Sambamba tools (v0.4.7) [23].
Somatic mutation detection and significantly mutated genes identification
Identification of somatic single-nucleotide variants (SNVs) was conducted by muTect (v 1.1.4) [24], and the somatic InDels were detected by Strelka (v1.0.13) [25]. ANNOVAR (ANNOVAR_2015Mar22) [26] was used to annotate variant call format files. The Mutational Significance in Cancer (MuSic, version: Genome-Model-Tools-Music-0.04) algorithm [27] was used to identify significantly mutated genes (SMGs) from the profiles of somatic SNVs and InDels (False discovery rate (FDR) < 0.25).
Mutation signature analysis
Mutational signatures were characterized according to the 96-substitution classification. Based on the frequency of 96 mutation types, Nonnegative Matrix Factorization (NMF) method (v0.22) was performed to extract mutational signatures and compare them with 30 known signatures referenced in the Catalogue of Somatic Mutations in Cancer (COSMIC) database using SomaticSignatures packages (v2.24.0) [28]. The similarity of mutation signatures was evaluated with cosnine similarity > 0.9, which suggested common signatures. Signatures 1, 6, 17, and 29 were identified in our samples. Then unsupervised hierarchical clustering was performed to identify the clusters of mutational signatures (Sig-cluster) according to the proportional contribution of each signature per sample.
Copy number analysis
Somatic copy number variations (SCNVs) were identified using CNVkit [29]. Then GISTIC 2.0 (v 2.0.22) [30] was used to identify the genome regions with significant alterations and screen out the recurrent CNV regions (parameters: -rx 0 -ext xls -fname ALL -ta 0.1 -td 0.1 -js 4 -qvt 0.25 -cap 1.5 -board 1-maxseg 2000 -conf 0.99 -genegistic 1 -armpeel 1 -brlen 0.7 -gcm extreme -savegene 1). In addition, unsupervised hierarchical clustering was performed to identify the clusters of copy-number variations (CNV-cluster) based on discretized CNVs.
HLA genotyping and neoantigen prediction
The raw data of WES were processed by software TSNAD (available on http://github.com/jiujiezz/TSNAD) [31]. This software developed by our research group combines multiple algorithms to identify somatic mutations, determine HLA genotyping, and predict neoantigens. After identification of somatic mutations, TSNAD can determine HLA genotyping by SOAP-HLA [32]. Subsequently, NetMHCpan [33] was invoked to predict mutation-derived neoantigens which could bind to class I MHC molecules. Then unsupervised hierarchical clustering was conducted to identify the neoantigen clusters (NEA-cluster) according to the neoantigen number. Since we lack expression data, there might be over-prediction of neoantigens to some extent.
Analysis of clonal architecture of somatic mutations
The R package SciClone [34] (http://github.com/genome/sciclone) was used to infer the clonal and subclonal architecture of somatic mutations by analyzing the variant allele frequencies in an individual sample using the Bayesian binomial mixture model. SciClone parameters: minimumDepth = 20, maximumClusters = 10, copyNumberMargins = 0.25. Variant allele frequencies were clustered and visualized as previously reported [34]. The clusters of clonality (Clonality-cluster) was defined according to the clonal patterns.
Integrative clustering
The consensus clustering was used to uncover molecular subtypes according to multidimensional genomic features, including Sig-cluster, CNV-cluster, NEA-cluster, Clonality-cluster, essential SMGs, and CNVs. Essential SMGs were selected with the overlap between the genes with a mutation frequency ≥ 10% in our cohort and the cancer-related genes in COSMIC [35] (https://cancer.sanger.ac.uk/cosmic/). Essential CNVs were selected with the overlap among the genes with alteration frequency ≥ 10%, the cancer-related genes in COSMIC and the genes involved in the key pathways in our cohort. The values of these variables are shown in the Table S1. Clustering was then done in R software (version 3.6.1) based on Euclidean distance using Ward's method.
Integrated pathway analysis
Geneset of cancer-driving genes from Cancer Gene Census (CGC) was accessed from the COSMIC. Subsequently, according to the genetic alteration rates of the cancer-driving genes in our cohort, we performed KEGG pathway enrichment analysis using DAVID bioinformatics Resource 6.8.
Immunohistochemical staining
In order to evaluate PD-L1 protein expression in gastric cancers, formalin-fixed, paraffin-embedded (FFPE) tissue sections were retrieved from tumor block of our GC cohort. 4um paraffin sections were routinely deparaffinized and rehydrated. Antigen retrieval was performed and endogenous peroxidase, non-specific protein binding sites were blocked and subsequently incubated in primary antibodies overnight at 4 ℃. PD-L1 immunohistochemical (IHC) was performed using rabbit anti-human PD-L1 (clone MXR003, MXB biotechnologies, Fujian China), which recognizes an epitope in the PD-L1 cytoplasmic domain and reactivity confirms the full-length PD-L1 protein expression. PD-L1 staining was observed in tumor cells and adjacent immune cells. Combined Positive Score (CPS) methods was introduced in our IHC study. Tumor cells, adjacent lymphocytes and phagocytes with positive membrane staining were counted as positive cells, and CPS ≥ 5 was annotated as positive staining (representatives were shown in Fig. S1) [36]. Observation and scoring was performed by an expert immunopathologist (Mei Kong).
Statistical analysis
The statistical analysis was performed by SPSS 21.0 software. The differences between categorical variables were compared by Pearson’s Chi-square test or Fisher’s exact test. The continuous or non-parametric variables were assessed by one-way ANOVA or Mann–Whitney test, respectively. Survival data were analyzed by Kaplan–Meier curves with log-rank test. Univariate and multivariate Cox regression analysis were performed to calculate the hazard ratio and 95% confidence interval. A P value of < 0.05 was considered significant.
Results
Patients’ clinicopathological features
Paired fresh-frozen tumor tissue and adjacent normal tissue were collected from a cohort of 70 pathologically confirmed GC patients (ZJU-GC) (Table S2). Among the patients, 47 (67.1%) were males and their median age at the time of diagnosis was 60 years (range 38–80 years old). The majority (61.4%) of the patients were diagnosed with TNM stage III or stage IV. Besides, 51.4% of cases were identified as intestinal-type and 57.1% were with poor differentiation. During the follow-up, 21.4% of cases developed peritoneal dissemination and 15.7% of cases developed liver metastasis.
Landscape of somatic mutations of GC
WES was conducted on 70 matched normal-tumor pairs from GC patients with a mean depth of 314- and 176-fold, respectively (Table S3). A total of 57,580 somatic mutations were identified from all samples (10,506 were of non-synonymous and 3560 were of synonymous) (Tables S4 and S5). In addition, 32 genes were identified as significantly mutated genes (SMGs) by the MuSiC algorithm (Fig. 1A). Of these, 14 were reported as SMGs in at least one previous GC genome sequencing study, such as TP53, ARID1A, PIK3CA, CDH1, RHOA, and SMAD4 (Table S6). However, 18 mutations have not been previously reported in GC samples (e.g., PREX2, PIEZO1, and FSIP2). Distribution of nonsynonymous somatic TP53 mutations is illustrated in Fig. 1B, and 87.2% occurred in a DNA-binding domain. The distribution of mutation sites for other three recurrent SMGs (CSMD3, LRP1B, and SYNE1) is also presented in Fig. S2. The mutational frequencies of the majority of SMGs were similar to those in TCGA-GC, whereas ARID1A and PIK3CA were mutated at remarkably lower frequencies in ZJU-GC (23% vs. 10% and 18% vs. 8%, Fig. 1C, Table S7), and the frequency of PIEZO1 mutation in ZJU-GC cohort was significantly higher than that in TCGA-GC (12% vs. 0%, P < 0.01). Notably, LRP1B was the more frequent mutant in intestinal type tumors (P = 0.03, Fig. 1D, Table S8) and tended to be less frequent in those with poor differentiation (P = 0.052). In addition, mutations of FAT4 were exclusive in intestinal type tumors (25% vs. 0%, P = 0.002).

The landscape of somatic mutations in ZJU-GC cohort. A Somatic mutations of 70 paired samples in ZJU-GC cohort. The middle matrix shows the somatic mutations by gene (row) and by sample (column). The top histogram shows the number of non-synonymous and synonymous mutations in each individual sample. The top tracks show the clinicopathological characteristics, including gender, age, Lauren types, differentiation, and TNM stage. The left histogram shows the number of somatic mutations accumulated on 70 ZJU-GC samples in each gene. The right histogram shows p values of each gene calculated from MuSic analysis. B Distribution of non-synonymous TP53 somatic mutations identified. C Comparison of gene mutation rate between ZJU-GC cohort and TCGA-GC. Orange or blue dots represent the genes with significantly higher or lower mutation rate, respectively. D Comparison of gene mutation rates by clinicopathological subtypes
KEGG pathway analysis and targetable genes
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of somatic mutations and somatic CNV revealed altered signaling pathways in GC, including RTK/RAS/PI(3)K, p53/cell cycle, cell adhesion, and chromatin remodeling (Fig. 2). The RTK/RAS/PI(3)K pathway was primarily altered by amplifications of ERBB2 (14%), HSP90AB1 (10%), and MYC (6%), as well as PIK3CA mutation (7%). P53/cell cycle pathway was frequently altered mainly owing to recurrent TP53 mutations (56%) and amplifications of cyclin-encoding genes (CCNE1 (10%), CCND1 (8%), and CCND3 (8%)). Other frequently altered genes included those involved in focal adhesion (RHOA, ITGAV), adherens junction (CDH1) or chromatin remodeling (ARID1A, SMARCA4, KMT2C, and KMT2D). In addition to the well-established target, ERBB2 amplification, other alterations, such as PIK3CA mutation and CDK4 amplification, could serve as potential therapeutic targets. According to the OncoKB database (https://www.oncokb.org), 31 out of 70 patients harbored at least one potentially targetable gene alteration (Table S9).

Genomic alterations of signaling pathways in ZJU-GC cohort. Gene somatic mutations and copy number variations are characterized in key signaling pathways, including RTK/RAS/PI(3)K pathway, cell adhesion pathway, cell cycle pathway, and chromatin remodeling pathway. Genes are grouped by the pathways and linked by the line and arrows showing molecular interactions
Identification and characterization of multidimensional genomic features
Mutational signatures
The predominant somatic mutation types were C:G > T:A transitions and C:G > A:T transversions (Fig. S3). A total of four independent mutational signatures, COSMIC Signature 1 (initiated by spontaneous deamination of 5-methylcytosine associating with aging [12]), Signature 6 (associated with deficient DNA mismatch repair [12, 37]), Signature 17 (attributed to oxidative stress [38] and associated with a poor prognosis [38]), and Signature 29 (as a tobacco-related signature [39]) could be identified (Fig. 3A, Table S10). Using unsupervised clustering, GC samples were divided into three subtypes (Sig-cluster1, Sig-cluster2, and Sig-cluster3), according to the proportional contribution of each signature per sample (Fig. 3B). We further investigated the association between these subtypes and OS. The Sig-cluster1 subtype had the longest OS, while Sig-cluster3 had the shortest (log-rank P < 0.001, Fig. 3C). Subsequently, multivariate Cox regression analyses also indicated a significant difference in prognosis among those subtypes (Fig. S4, Table S11).

Characterization and identification of genomic features including mutational signatures (A–C), copy number variations (D–F), predicted neoantigen (G–J), and clonality (K–N) in ZJU-GC cohort. A Mutational signatures are characterized according to the 96-substitution classification, with horizontal axis showing mutation types of 96 substitutions and vertical axis showing the estimated mutations of each mutation type. B Unsupervised clustering of mutational signatures for 70 GC samples. C Association between three clusters of mutational signatures and OS in ZJU-GC cohort. D GISTIC 2.0 significant CNVs with amplifications on left and deletions on right. E Unsupervised clustering of CNVs for 70 GC samples. F Association between three clusters of CNVs and OS. G Predicted combining site of neoantigen. H Correlation between TMB and TNB in ZJU-GC cohort. I Unsupervised clustering of predicted neoantigens and somatic mutations in 70 ZJU-GC samples. The middle matrix shows the predicted neoantigens and somatic mutations by gene (row) and by sample (column). The top histograms show TNB and TMB. The right histogram shows the number of predicted neoantigens and somatic mutations accumulated on 70 GC samples in each gene. According to the status of predicted neoantigens, 70 samples are divided into two clusters. J Comparison of TNB between NEA-cluster 1 and NEA-cluster 2. K Identification of tumor clonality. L Classification of tumor clonality in ZJU-GC. According to the clonal status, 70 samples are divided into two clonality groups: oligoclonal group and multiclonal group. M Comparison of histological characteristics between oligoclonal group and multiclonal group, including differentiations, Lauren type, and PD-L1 status. N Comparison of TMB between oligoclonal group and multiclonal group. Sig-cluster: mutational signature cluster; CNV-cluster: copy number variation cluster; NEA-cluster: neoantigen cluster; TMB: tumor mutation burden; TNB: tumor neoantigen burden; OS: overall survival. ***P < 0.001, **** P <0.0001
Somatic CNVs
Forty-two significant focal CNVs (24 with amplification and 18 with deletion) were identified using GISTIC 2.0 (Fig. 3D, Table S12). The recurrent CNVs with amplification included CCNE1 (19q12), ERBB2 (17q12), CCND1 (11q13.3), MUC4 (3q29), VEGFA (6q21.1), and NOTCH1 (9q34.3). Significantly deleted CNVs included 1p35.3, 1p21.1, 3p21.2, 9p21.3, 16q21, 17q12, and 19p13.3, encompassing a great number of tumor suppressor genes, including ARID1A, MLH1, CDKN2A, CDH1, BRCA1, TP53, and SMARCA4. Based on significantly altered CNVs per sample, 70 GC patients were clustered into three subtypes by unsupervised clustering (CNV-cluster1, CNV-cluster2, and CNV-cluster3; Fig. 3E). We further observed that the three subtypes were significantly interrelated to prognosis (P = 0.038, Fig. 3F) and associated with the patients’ age at diagnosis, gender, and the Lauren subtype (Table S13).
Predicted neoantigens derived from mutated genes
A total of 4994 predicted neoantigens were identified in 70 patients (Table S14). The number of predicted neoantigens ranged from 0 to 918 (median, 52). Besides, there were 873 (17.5%) neoantigen affinities to allele HLA-B58:01 (Fig. 3G, Table S15). The number of neoantigens was significantly correlated with tumor mutational burden (TMB) (R2 = 0.879, P < 0.001, Fig. 3H). Neoantigens were frequently derived from somatic mutations of TTN (13%), followed by CSMD3 (10%) (Table S16). Unsupervised clustering according to predicted neoantigens categorized GC into two clusters (Fig. 3I) with significantly different tumor neoantigen burden (TNB) (P < 0.001, Fig. 3J). The associations between these two clusters and clinicopathological characteristics are presented in Table S17 and Fig. S5.
Clonal and subclonal architectures
SciClone was applied to specimens to reconstruct the clonal and subclonal architectures, reflecting the intratumor heterogeneity. Patterns of clonality included monoclonal (a single dominant clone with or without one minor subclone), biclonal (two major clones), and complex clonal patterns (more than two clones (Fig. 3K)). We defined GC with monoclonal and biclonal patterns as oligoclonal and those with complex clonal pattern as multiclonal (Fig. 3L). Comparison of clinicopathological features of these two subtypes showed that multiclonality was significantly associated with well or moderate differentiation, the Lauren intestinal type, and programmed death-ligand 1 (PD-L1) positivity (Fig. 3M and Table S18). GC cases with multiclonal features carried significantly higher TMB (Fig. 3N).
Integrative genomic classification of GC
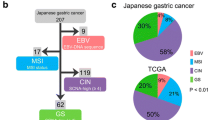
To identify genomic classification of GC associated with patients’ clinicopathological features and survival outcomes, unsupervised clustering was conducted based on subgroups derived from mutational signatures, CNVs, predicted neoantigens, clonality, as well as essential gene alterations. As shown in Fig. 4A, 70 GC cases were divided into four subtypes (ZJU-GC subtypes): subtype 1 (n = 22), subtype 2 (n = 16), subtype 3 (n = 12), and subtype 4 (n = 20). Subsequently, we investigated the association of the four subtypes with OS (Fig. 4B). We found that subtype 4 had the longest OS, while subtype 2 and subtype 3 both had significantly the shortest OS than subtype 4, and subtype 1 had a moderate OS. Thereafter, associations between the four subtypes and TCGA molecular subtypes or Lauren histological types were further explored. We observed that 90.9% of tumors in subtype 1 were classified into CIN subtype, whereas subtype 2 contained 50.0% CIN type and 50.0% GS type (Fig. 4C, Table S19). The majority of tumors in subtype 3 (83.3%) and subtype 4 (75.0%) were classified into GS type. Notably, GC tumors in subtype 1 and subtype 2 had a significantly higher proportion of intestinal histology compared with subtype 3 and subtype 4 (P < 0.01, Fig. 4D). Furthermore, when examining the first site of metastasis, we found that subtype 1 harbored the highest rate of liver metastasis in comparison with other subtypes (P < 0.01), whereas subtype 3 and subtype 4 developed significantly higher rate of peritoneal metastasis in comparison with other subtypes (P < 0.05) (Fig. 4E, Fig. S6). In addition, those in subtype 1 and subtype 2 harbored higher TMB compared with those in the other two subtypes, as well as higher TNB than that in subtype 4 (Fig. 4F). To further explore the prognostic value of the proposed classification system, multivariate Cox regression analysis was undertaken, and it was found that the classification system was an independent prognostic factor after adjusting for TNM stage and TMB (Fig. 4G). Four subtypes displayed distinct molecular features (Fig. 4H, Table S20). Subtype 1 harbored recurrent mutations in TP53 (81.8%), as well as amplification of ERBB2 (31.8%) and HSP90AB1 (27.3%). Subtype 2 was enriched in frequent mutations of TP53 (87.5%), LRP1B (56.3%), and SYNE1 (56.3%). Subtype 3 and subtype 4 both exhibited frequent deletion of ARID1A (subtype 3, 16.7%; subtype 4, 20.0%).

Integrated genomic classification of gastric cancer in association with clinicopathological features and patient outcomes. A Unsupervised clustering of integrated genomic features (Sig-cluster, CNV-cluster, NEA-cluster, clonality-cluster), frequent mutated genes, and copy number variations. 70 GC samples are divided into four ZJU-GC subtypes: subtype 1 (blue), subtype 2 (brown), subtype 3 (rose red), and subtype 4 (green). Clinicopathological and molecular characteristics are depicted at the bottom. B Association between ZJU-GC subtypes and OS. C Sankey diagram showing the association between ZJU-GC subtypes and TCGA subtypes. D Association between ZJU-GC subtypes and Lauren types. E Comparison of first-metastasis site among ZJU-GC subtypes. F Comparison of TMB and TNB among ZJU-GC subtypes. G Forest plot showing univariate and multivariate Cox regression analysis for the association between ZJU-GC subtype and OS. H Molecular characteristics of four ZJU-GC subtypes. Sig-cluster: mutational signature cluster; CNV-cluster: copy number variation cluster; NEA-cluster: neoantigen cluster; TMB: tumor mutation burden; TNB: tumor neoantigen burden; OS: overall survival; CIN: chromosomal instability; EBV: Epstein-Barr virus; MSI: microsatellite instability; GS: genomically stable. * P < 0.05
In summary, we proposed a new genomic classification system, including four subtypes associated with distinct metastatic patterns and prognosis (Fig. 5). Subtype 1, which is predominantly Lauren intestinal type, harbored recurrent TP53 mutation and ERBB2 amplification, TMB/ TNB, intratumoral heterogeneity, and has a liver metastasis tendency. Subtype 2 tends to occur at an elder age, accompanying with high TP53 and SYNE1 mutations, high TMB/TNB, and is associated with poor prognosis. Subtype 3 and subtype 4 include GC patients with mainly diffuse/mixed type tumors, high frequency of peritoneal metastasis, and genomical/chromosomal stability, whereas subtype 4 is associated with a favorable prognosis.

Summary of key features of gastric cancer in four genomic subtypes. The schematic shows the salient characteristics associated with each of four ZJU-GC subtypes
Independent cohort validation of the genomic classification
In order to increase the clinical applicability of our genomic classification system, we performed dimensionality reduction analysis, and five features (Sig-cluster, CNV-cluster, FAT4 mutation, LRP1B mutation, and CCNE1 CNV) were selected. Then we constructed a simplified genomic classification model to predict subtypes based on naive Bayes algorithm (details were shown in supplementary methods and Fig. S7). Tenfold cross-validation of this prediction model was performed and internal validation accuracy was up to 95.7%.
WES was performed in 23 GC tumors and paired normal gastric mucosa from an independent cohort (supplementary methods and Table S21–22). After genomic classification prediction using the model based on five selected genomic features, we achieved predicted ZJU-GC subtype for each sample in the validation cohort (Table S22). 23 GC cases were divided into four subtypes: subtype 1 (n = 10), subtype 2 (n = 5), subtype 3 (n = 3), and subtype 4 (n = 5). Similar to ZJU-GC cohort, the genomic subtype showed significant association with metastasis pattern in the validation cohort that subtype 1 harbored the highest rate of liver metastasis while subtype 3 and subtype 4 showed higher rate of peritoneal metastasis (Fig. S8A-B). Further analysis revealed that GC cases of subtype 1 had higher prevalence of Lauren intestinal histology and those of subtype 3 and subtype 4 were enriched in diffuse/mixed type (Fig. S8C). In addition, subtype 1 and subtype 2 harbored higher TMB and SYNE1 mutation compared with the other two subtypes. Subtype 1 was enriched in TP53 mutation and ERBB2 amplification (Fig. S8D–E). Therefore, our validation suggested that the molecular classification had good expansibility and robustness and confirmed the relevance between genomic subtypes and clinicopathological features.
Discussion
In the current study, we characterized the landscape of genomic alterations in Chinese GC patients. Based on mutational signature, CNV, predicted neoantigen, clonality analysis, and essential genomic alterations, we presented four subtypes of GC associated with different clinical phenotypes. The current study added further layers of information about the genomic characteristics and molecular classification of GC and could lead to improvement in patient stratification, prognostication, and genome-guided therapy.
Based on the results of WES, we identified the SMGs in the current cohort. Among 32 SMGs identified, 14 (e.g., TP53, SPTA1) were reported as SMGs in at least one of 9 previous GC-based studies [40,41,42,43,44,45,46,47,48], while 18 genes, such as PREX2, PIEZO1, TRHDE, FSIP2, and FAT3, have not been previously reported. Among them, the most frequently mutated gene was TP53 in the current study, which encoded the tumor suppressor and transcription factor p53. Previous genomic-based studies confirmed the high frequency of TP53 mutations in GC samples (40–50%) [8, 40], which is consistent with the present study (56%). To date, no drug abrogating the oncogenic functions of p53 mutant has been approved for the treatment of cancer [49]. A recent study suggested the potential of arsenic trioxide in treating p53-mutated cancer patients [50]. Importantly, in the current research, 2 ZJU-GC subtypes (subtypes 1 and 2) were enriched for TP53 mutations (81.8% and 87.5%, respectively, Fig. S9), suggesting that they may benefit from TP53-targeted therapy. Spectrin Alpha, Erythrocytic 1 (SPTA1) encodes a member of a family of molecular scaffold proteins, linking the plasma membrane to the actin cytoskeleton. SPTA1 mutation is associated with a variety of hereditary red blood cell disorders, such as hereditary elliptocytosis and hereditary spherocytosis [51]. In the present study, SPTA1 was identified as a SMG for GC, which was consistent with two previous genomic-based studies [42, 45]. In addition, in a recent study on Mongolians with hepatocellular carcinoma, SPTA1 was reported as a potential driver gene [52]. However, whether SPTA1 is a driver gene in GC and how SPTA1 mutation mechanistically contributes to tumorigenesis remained elusive. Phosphatidylinositol-3,4,5-Trisphosphate Dependent Rac Exchange factor 2 (PREX2) belongs to PREX family and is an important regulator of Purkinje cell morphogenesis and motor coordination [53]. Although PREX2 was not reported as a SMG or a driver gene in previous GC-based studies, it was reported as a SMG in a study on melanoma [54], as well as being a candidate driver gene of pancreatic carcinogenesis [55]. A recent study on 46 patients with advanced GC who received anti-programmed cell death protein 1 (PD-1) antibody revealed that PREX2 mutation was correlated with a poor progression-free survival [56]. Understanding the role of key mutations in GC tumorigenesis and progression are promising aspects in future research.
In the decision tree of TCGA classification derived from analysis of six molecular platforms, DNA-level features, such as EBV, MSI, and the degree of SCNVs could substantially differentiate into four distinct subgroups [7]. Li et al. identified two molecular subtypes of non-hypermutated GC that correlated with prognosis by SMG analysis [57]. To the best of our knowledge, we first proposed an integrated genome-based classification system, including mutational signature, clonality, and neoantigen features. Mutational signatures have been attributed to specific carcinogenic factors with clinical relevance in multiple types of cancer [12, 58,59,60]. Clonality, which reflects intratumor heterogeneity, is associated with cancer aggressiveness, treatment resistance, and patient prognosis [13, 44]. Tumor-specific neoantigen, derived from accumulation of somatic mutations, has been proposed as a predictive indicator for response to immune checkpoint inhibitors [61,62,63,64].
We proposed a novel genome-based classification system with significant clinical relevance and prognostic value. Subtype 4 was found to be associated with significantly longer OS than the other subtypes, whereas subtype 2 and subtype 3 showed a relatively poorer prognosis, and subtype 1 had a moderate prognosis. In the TCGA classification system, studies showed that EBV and MSI had a better prognosis [65, 66], while the difference in OS between GS and CIN subtypes was found inconsistent in different cohorts [8, 67]. Compared with the original prognosis trend of our classification system, classifying ZJU-GC cohort using the TCGA genomic scheme yielded a weaker association with the prognosis, where GS and CIN subtypes showed no significant difference in OS (Fig. S10), which is similar to the result of ACRG cohort [8]. Tumors in subtype 1 were mainly grouped into CIN type, while those in subtype 3 and subtype 4 were grouped into GS subtype. Notably, subtype 3 and subtype 4 showed a remarkable difference in OS in the present analysis; similar result was found between patients with peritoneal metastasis in the two subgroups (Fig. S11). In molecular level, the distinction of these two subtypes was mainly attributed to the distribution of mutational signatures, i.e., enrichment of signature 1 (known as clock-like mutational process) in subtype 4. Signature 1 is commonly found in various types of cancer and is associated with patients’ age at diagnosis [68], and it has been reported to be a negative prognostic factor in triple-negative breast cancer [69], while its clinical relevance in GC has not been reported yet. Multiple difference has also been found significant CNV between subtypes 3 and 4, and subtype 4 harbored significant amplifications in more regions than subtype 3 (Fig. S12, Table S23). The proportion of GS tumors in our cohort (49%) and another GC cohort in Asia (45%) [8] was higher than that in TCGA (24%). Differentiation of subtype 3 and subtype 4 could be clinically informative as it reflects heterogeneity within GS subtype in Chinese patients, though validation in larger cohort is required. Multiomic analysis might further suggest the underlying mechanism of the prognosis difference. The immunity features of the four subtypes were also described in the current study. Subtype 1 and subtype 2 harbored higher TMB/TNB, PD-L1 expression, as well as more frequently LRP1B and SYNE1 mutations. These findings indicated that the two mentioned subtypes may have a better response to immune checkpoint inhibitors [62, 64, 70,71,72]. Nevertheless, further studies with larger sample size need to be carried out to confirm our findings.
Another key finding of this study is that we identified GC subtypes associated with patterns of metastasis in synchronous or metachronous metastatic GC, and this relevance was reproduced in the validation cohort. Peritoneal dissemination and liver metastasis are the major metastatic patterns of GC. To our knowledge, information on metastasis pattern has not been reported by TCGA-GC classification. In the present research, we examined the first site of metastasis regardless of surgical resection, and observed a significant tendency of liver metastasis in subtype 1, as well as a tendency of peritoneal metastasis in subtype 3 and subtype 4. Lee et al. reported that diffuse/mixed type was associated with a peritoneal recurrence, and the Lauren intestinal type was associated with hematogenous metastasis [73]. In the current study, in both ZJU-GC cohort and the validation cohort, subtype 1 was enriched in the Lauren intestinal type, and subtype 3 and subtype 4 were enriched in diffuse/mixed type. Among Koreans, the ACRG research also indicated the existence of subtypes with different metastatic tendency based on transcriptomic analysis [8]. The current study, for the first time, proposed a genomic classification that differentiates molecular subtypes associated with tendency to liver or peritoneal metastasis. On the molecular basis, subtype 1 had the highest prevalence of HSP90AB1 and ERBB2 amplifications among the four subtypes. HSP90AB1 was previously found to promote epithelial-mesenchymal transition via Wnt/β-catenin signaling pathway in GC progression [74]. A large-scale prospective study reported that ERBB2 positivity was significantly correlated to liver metastasis and absence of peritoneal metastasis [75]. Additionally, deletion of ARID1A was commonly found in subtype 3 and subtype 4, and it was revealed that reduced expression of the chromatin remodeling gene could promote migration and invasion of GC cells by downregulating E-cadherin expression [76]. Collectively, the proposed classification system can be informative to guide studies on molecular oncology. Nevertheless, the underlying mechanism of different site-specific patterns of metastasis among ZJU-GC subtypes requires further investigation through in vitro and in vivo experiments. Furthermore, our classification could also guide site-specific recurrence monitoring and treatments in clinical scenario. For instance, an evidence suggested that addition of pre-emptive intraperitoneal chemotherapy to gastrectomy reduces the risk of peritoneal recurrence for GC patients who are at high-risk of peritoneal metastasis [77, 78]. Moreover, an ongoing phase III trial is currently recruiting GC patients at high risk of peritoneal metastasis (T3/4, irrespective of nodal or peritoneal cytology status) [79]. The combination of clinicopathological factors and the proposed genomic classification system may lead to a better risk stratification of peritoneal metastasis.
There are some limitations in the current study that should be clearly declared. This is a single-center, retrospective study and the sample size is relatively limited. In addition, in spite of an obvious clinical significance of the newly proposed genomic classification, little is known about its underlying mechanism. Therefore, future prospective, multi-center, and large cohort studies are warranted to confirm our findings and to search for the molecular basis of the different clinical phenotypes (e.g., metastatic pattern). Although the proposed classification system is clinically significant, there are still obstacles towards broad clinical application due to the lack of large cohort validation. Further optimization of the classification algorithm and large cohort validation will be the next directions.
In summary, the present study revealed the genomic characteristics of Chinese GC patients and proposed a unique genomic classification system with significant clinical relevance. Through interpretation of genomic information, this study provided a rationale for research on GC and for molecular classification of multiple types of cancer to guide precision medicine.
References
Thrift AP, El-Serag HB. Burden of gastric cancer. Clin Gastroenterol Hepatol. 2020;18(3):534–42.
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–32.
Yang L, Zheng R, Wang N, Yuan Y, Liu S, Li H, Zhang S, Zeng H, Chen W. Incidence and mortality of stomach cancer in China, 2014. Chin J Cancer Res. 2018;30(3):291–8.
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396(10251):635–48.
Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24(9):1449–58.
Pectasides E, Stachler MD, Derks S, Liu Y, Maron S, Islam M, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov. 2018;8(1):37–48.
Cancer Genome Atlas Research. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9.
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21(5):449–56.
Lei Z, Tan IB, Das K, Deng N, Zouridis H, Pattison S, et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology. 2013;145(3):554–65.
Hollis RL, Thomson JP, Stanley B, Churchman M, Meynert AM, Rye T, et al. Molecular stratification of endometrioid ovarian carcinoma predicts clinical outcome. Nat Commun. 2020;11(1):4995.
Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol. 2018;68(5):959–69.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613–28.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74.
Liu XS, Mardis ER. Applications of immunogenomics to cancer. Cell. 2017;168(4):600–12.
De Mattos-Arruda L, Vazquez M, Finotello F, Lepore R, Porta E, Hundal J, et al. Neoantigen prediction and computational perspectives towards clinical benefit: recommendations from the ESMO Precision Medicine Working Group. Ann Oncol. 2020;31(8):978–90.
Zhou J, Zhao W, Wu J, Lu J, Ding Y, Wu S, et al. Neoantigens derived from recurrently mutated genes as potential immunotherapy targets for gastric cancer. Biomed Res Int. 2019;2019:8103142.
Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol. 2006;12(3):354–62.
Serra O, Galan M, Ginesta MM, Calvo M, Sala N, Salazar R. Comparison and applicability of molecular classifications for gastric cancer. Cancer Treat Rev. 2019;77:29–34.
Lin SJ, Gagnon-Bartsch JA, Tan IB, Earle S, Ruff L, Pettinger K, et al. Signatures of tumour immunity distinguish Asian and non-Asian gastric adenocarcinomas. Gut. 2015;64(11):1721–31.
Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, et al. The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. Cancer J Clin. 2017;67(2):93–9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9.
Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–7.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22(8):1589–98.
Gehring JS, Fischer B, Lawrence M, Huber W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015;31(22):3.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12(4):e1004873.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41.
Zhou Z, Lyu X, Wu J, Yang X, Wu S, Zhou J, Gu X, Su Z, Chen S. TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection. Royal Soc Open Sci. 2017;4(4):170050.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7.
Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2008;61(1):1–13.
Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014;10(8):e1003665.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941–7.
Derks S, Liao X, Chiaravalli AM, Xu X, Camargo MC, Solcia E, et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget. 2016;7(22):32925–32.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073-87.e3.
Lin Y, Luo Y, Sun Y, Guo W, Zhao X, Xi Y, et al. Genomic and transcriptomic alterations associated with drug vulnerabilities and prognosis in adenocarcinoma at the gastroesophageal junction. Nat Commun. 2020;11(1):6091.
India Project Team of the International Cancer Genome Consortium. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. 2013;4:2873.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9.
Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43(12):1219–23.
Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44(5):570–4.
Liu J, McCleland M, Stawiski EW, Gnad F, Mayba O, Haverty PM, et al. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat Commun. 2014;5:3830.
Chen K, Yang D, Li X, Sun B, Song F, Cao W, et al. Mutational landscape of gastric adenocarcinoma in Chinese: implications for prognosis and therapy. Proc Natl Acad Sci USA. 2015;112(4):1107–12.
Park H, Cho SY, Kim H, Na D, Han JY, Chae J, et al. Genomic alterations in BCL2L1 and DLC1 contribute to drug sensitivity in gastric cancer. Proc Natl Acad Sci USA. 2015;112(40):12492–7.
Cho SY, Park JW, Liu Y, Park YS, Kim JH, Yang H, et al. Sporadic early-onset diffuse gastric cancers have high frequency of somatic CDH1 alterations, but low frequency of somatic RHOA mutations compared with late-onset cancers. Gastroenterology. 2017;153(2):536–49.
Cong T, Liu GX, Cui JX, Zhang KC, Chen ZD, Chen L, Wei B, Huang XH. Exome sequencing of gastric cancers screened the differences of clinicopathological phenotypes between the mutant and the wide-type of frequently mutated genes. Zhonghua Yi Xue Za Zhi. 2018;98(28):2242–5.
Chen C, Shi C, Huang X, Zheng J, Zhu Z, Li Q, et al. Molecular profiles and metastasis markers in Chinese patients with gastric carcinoma. Sci Rep. 2019;9(1):13995.
Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018;15(1):13–30.
Chen S, Wu JL, Liang Y, Tang YG, Song HX, Wu LL et al: Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2020.
Maillet P, Alloisio N, Morlé L, Delaunay J. Spectrin mutations in hereditary elliptocytosis and hereditary spherocytosis. Hum Mutat. 1996;8(2):97–107.
Candia J, Bayarsaikhan E, Tandon M, Budhu A, Forgues M, Tovuu LO, et al. The genomic landscape of Mongolian hepatocellular carcinoma. Nat Commun. 2020;11(1):4383.
Srijakotre N, Man J, Ooms LM, Lucato CM, Ellisdon AM, Mitchell CA. P-Rex1 and P-Rex2 RacGEFs and cancer. Biochem Soc Trans. 2017;45(4):963–77.
Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485(7399):502–6.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Jin Y, Chen DL, Wang F, Yang CP, Chen XX, You JQ, et al. The predicting role of circulating tumor DNA landscape in gastric cancer patients treated with immune checkpoint inhibitors. Mol Cancer. 2020;19(1):154.
Li X, Wu WK, Xing R, Wong SH, Liu Y, Fang X, et al. Distinct subtypes of gastric cancer defined by molecular characterization include novel mutational signatures with prognostic capability. Cancer Res. 2016;76(7):1724–32.
Kan Z, Ding Y, Kim J, Jung HH, Chung W, Lal S, et al. Multi-omics profiling of younger Asian breast cancers reveals distinctive molecular signatures. Nat Commun. 2018;9(1):1725.
Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15(9):585–98.
Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017;7(10):1116–35.
Lu T, Wang S, Xu L, Zhou Q, Singla N, Gao J, et al. Tumor neoantigenicity assessment with CSiN score incorporates clonality and immunogenicity to predict immunotherapy outcomes. Sci Immunol. 2020;5(44):eazz3199.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–9.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Moehler M SK, Garrido M, Salman P, Shen L, Wyrwicz, et al: Nivolumab plus chemotherapy versus chemotherapy as first-line treatment for advanced gastric cancer/gastroesophageal junction cancer/esophageal adenocarcinoma: first results of the CheckMate 649 study. ESMO 2020, LBA6.
Camargo MC, Kim WH, Chiaravalli AM, Kim KM, Corvalan AH, Matsuo K, et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: an international pooled analysis. Gut. 2014;63(2):236–43.
Zhu L, Li Z, Wang Y, Zhang C, Liu Y, Qu X. Microsatellite instability and survival in gastric cancer: a systematic review and meta-analysis. Mol Clin Oncol. 2015;3(3):699–705.
Sohn BH, Hwang JE, Jang HJ, Lee HS, Oh SC, Shim JJ, et al. Clinical significance of four molecular subtypes of gastric cancer identified by The Cancer Genome Atlas Project. Clin Cancer Res. 2017. https://doi.org/10.1158/1078-0432.CCR-16-2211.
Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47(12):1402–7.
Chen H, Chong W, Yang X, Zhang Y, Sang S, Li X, Lu M. Age-related mutational signature negatively associated with immune activity and survival outcome in triple-negative breast cancer. Oncoimmunology. 2020;9(1):1788252.
Chen H, Chong W, Wu Q, Yao Y, Mao M, Wang X. Association of LRP1B mutation with tumor mutation burden and outcomes in melanoma and non-small cell lung cancer patients treated with immune check-point blockades. Front Immunol. 2019;10:1113.
Li P, Xiao J, Zhou B, Wei J, Luo J, Chen W. SYNE1 mutation may enhance the response to immune checkpoint blockade therapy in clear cell renal cell carcinoma patients. Aging. 2020;12(19):19316–24.
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–6.
Lee JH, Chang KK, Yoon C, Tang LH, Strong VE, Yoon SS. Lauren histologic type is the most important factor associated with pattern of recurrence following resection of gastric adenocarcinoma. Ann Surg. 2018;267(1):105–13.
Wang H, Deng G, Ai M, Xu Z, Mou T, Yu J, Liu H, Wang S, Li G. Hsp90ab1 stabilizes LRP5 to promote epithelial-mesenchymal transition via activating of AKT and Wnt/beta-catenin signaling pathways in gastric cancer progression. Oncogene. 2019;38(9):1489–507.
Matsusaka S, Nashimoto A, Nishikawa K, Miki A, Miwa H, Yamaguchi K, et al. Clinicopathological factors associated with HER2 status in gastric cancer: results from a prospective multicenter observational cohort study in a Japanese population (JFMC44-1101). Gastric Cancer. 2016;19(3):839–51.
Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ, Sun YH, Yang PY, Liu F. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis. 2014;35(4):867–76.
Cheong JH, Shen JY, Song CS, Hyung WJ, Shen JG, Choi SH, Noh SH. Early postoperative intraperitoneal chemotherapy following cytoreductive surgery in patients with very advanced gastric cancer. Ann Surg Oncol. 2007;14(1):61–8.
Desiderio J, Chao J, Melstrom L, Warner S, Tozzi F, Fong Y, Parisi A, Woo Y. The 30-year experience—a meta-analysis of randomised and high-quality non-randomised studies of hyperthermic intraperitoneal chemotherapy in the treatment of gastric cancer. Eur J Cancer. 2017;79:1–14.
Glehen O, Passot G, Villeneuve L, Vaudoyer D, Bin-Dorel S, Boschetti G, Piaton E, Garofalo A. GASTRICHIP: D2 resection and hyperthermic intraperitoneal chemotherapy in locally advanced gastric carcinoma: a randomized and multicenter phase III study. BMC Cancer. 2014;14:183.
Acknowledgements
The authors thank Zheng Ju and Hong Su (from Novogene Bioinformatics Technology Co. Ltd., Beijing, China) for assistance with bioinformatics analyses. The authors greatly thank all the staffs and assistants in the Department of Surgical Oncology, and Liming Xu in the Department of Pathology, the First Affiliated Hospital, Zhejiang University School of Medicine for their support in sample collection and immunohistochemistry study. This study was supported by Key Research and Development Program of Science and Technology Department of Zhejiang Province (No. 2018C03022, 2020C03112), Project supported by National Natural Science Foundation of China (No. 81803107), National Health and Family Planning Commission Research Fund & Zhejiang Provincial Medical and Health Major Science and Technology Plan Project (No. KWJ-ZJ-1802), Alibaba-Zhejiang University Joint Research Center of Future Digital Healthcare, Project of the Regional Diagnosis and Treatment Centre of the Health Planning Committee (No. JBZX-201903).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Ding, Y., Chen, Y. et al. A novel genomic classification system of gastric cancer via integrating multidimensional genomic characteristics. Gastric Cancer 24, 1227–1241 (2021). https://doi.org/10.1007/s10120-021-01201-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-021-01201-9