
Abstract
Although gastrointestinal stromal tumors (GISTs) are a rare type of cancer, they are the commonest sarcoma in the gastrointestinal tract. Molecularly targeted therapy, such as imatinib therapy, has revolutionized the treatment of advanced GIST and facilitates scientific research on GIST. Nevertheless, surgery remains a mainstay of treatment to obtain a permanent cure for GIST even in the era of targeted therapy. Many GIST guidelines have been published to guide the diagnosis and treatment of the disease. We review current versions of GIST guidelines published by the National Comprehensive Cancer Network, by the European Society for Medical Oncology, and in Japan. All clinical practice guidelines for GIST include recommendations based on evidence as well as on expert consensus. Most of the content is very similar, as represented by the following examples: GIST is a heterogeneous disease that may have mutations in KIT, PDGFRA, HRAS, NRAS, BRAF, NF1, or the succinate dehydrogenase complex, and these subsets of tumors have several distinctive features. Although there are some minor differences among the guidelines—for example, in the dose of imatinib recommended for exon 9-mutated GIST or the efficacy of antigen retrieval via immunohistochemistry—their common objectives regarding diagnosis and treatment are not only to improve the diagnosis of GIST and the prognosis of patients but also to control medical costs. This review describes the current standard diagnosis, treatment, and follow-up of GISTs based on the recommendations of several guidelines and expert consensus.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumors (GISTs) are considered potentially malignant tumors and are the commonest mesenchymal tumors in the gastrointestinal tract. Since the discovery of gain-of-function mutations in the KIT and PDGFRA genes and clinical application of tyrosine kinase inhibitors (TKIs), such as imatinib, our understanding of the molecular and clinical features of GISTs has increased substantially, and the diagnosis and treatment of GIST have rapidly and dramatically changed [1, 2]. These advances provided information that facilitated the preparation of clinical practice guidelines by the National Comprehensive Cancer Network (NCCN) [3] and the European Society for Medical Oncology (ESMO) [4]. Since the first guidelines were published, they have been updated annually or biannually, and other countries have published their own GIST guidelines [5–7]. It is suggested that diagnosis and treatment based on the guidelines will improve the prognosis of patients and the quality of medical care, as well as control medical costs. Last year, the Japanese and ESMO guidelines were updated, and there were consensus meetings of experts in several East Asian countries, including Japan, Korea, Taiwan, and China. This review discusses the current standard diagnosis, treatment, and follow-up of GISTs based on the guidelines and expert consensus [3–7].
Epidemiology and incidence
The worldwide incidence and prevalence of GIST are estimated to be approximately 1–1.5 per 100,000 per year and 13 per 100,000, respectively [8]. A recent report suggested that, except for incidental GIST, the age-adjusted incidence of clinical GIST was 0.8 per 100,000 per year on the basis of the data from the Surveillance, Epidemiology, and End Results program of the National Cancer Institute [9]. Population-based studies have shown that the median age at diagnosis is in the 60s, although GIST has been detected in all age groups. There is no significant sex difference. GIST in children and young adults, although rare, is a distinct subset of pediatric GIST, and syndromic GISTs may be found in children and individuals in early middle age [10, 11]. The predominant localization of GISTs seems to be the stomach (60 %) and small intestine (30–20 %), but GISTs may develop in the colorectum, esophagus, and, rarely, in the mesentery, omentum, or retroperitoneum (extragastrointestinal GIST), where KIT-positive mesenchymal cells are found.
Diagnosis
Clinical presentation
Many GISTs may be identified clinically because of symptoms including gastrointestinal bleeding and subsequent anemia, early satiety, abdominal distension, and discomfort and/or pain due to tumor compression [11]. However, GISTs are sometimes asymptomatic until advanced stages because of a submucosal localization and noninvasive behavior compared with carcinomas. Gastrointestinal examinations, including endoscopy, sometimes reveal asymptomatic GISTs, especially in the stomach. Hence, cancer-screening health examinations may increase the detection of asymptomatic GIST in the stomach [12]. GIST rarely metastasizes to lymph nodes, except for a special subtype of SDH-mutated GIST [10], and its spread to the extra-abdominal organs is extremely rare as an initial metastatic presentation.
The natural history of GIST remains largely unknown. Pathology reports on subclinical GISTs have shown an unexpectedly high incidence of microscopic GISTs in the stomach and small intestine [13, 14]. Small GISTs (from a few millimeters to less than 10 mm in diameter) are also commonly found in the proximal stomach of individuals older than 50 years. Immunohistochemistry reveals that these mini-GISTs are KIT-positive, and they often have an oncogenic mutation in the KIT or PDGFRA gene [15]. Most mini-GISTs are thought to be biologically indolent and do not progress during follow-up unless they have high-risk features such as an irregular border, internal heterogeneity, or ulceration [3, 16]. Although complete surgical resection is the mainstay of treatment for clinical and/or symptomatic GISTs, the clinical significance of surgical treatment remains unknown for asymptomatic and incidentally found mini-GISTs.
Pathological diagnosis, including rare GISTs
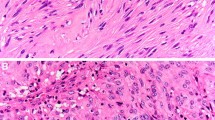
The pathological diagnosis of GIST depends on the morphology and immunohistochemical findings. The morphological features include a predominantly spindle cell type (70 %), epithelioid cell type (20 %), or mixed type (10 %). In addition, 95 % of GISTs are positive for KIT (CD117) and/or discovered on GIST-1 (DOG1), and 70 % are found to be positive for CD34 by immunohistochemistry. KIT positivity is a major defining feature for the diagnosis of GIST for a tumor that has morphological features compatible with GIST, although KIT positivity alone is not sufficient for the diagnosis (Fig. 1). When there is KIT negativity, as in approximately 5 % of GISTs, DOG1 staining, followed by CD34 staining, is considered diagnostic. The other important molecular marker that is useful in the diagnosis of GISTs is the presence of mutations in either KIT or PDGFRA; nearly 80 % and 10 % of GISTs, respectively, are positive for these mutations. When gastric GISTs have no mutations in KIT or PDGFRA, immunostaining for succinate dehydrogenase (SDH) iron–sulfur subunit (subunit B) (SDHB) is recommended [10]. The mitotic count is of prognostic value and should be expressed as the number of mitoses for a total area of 5 mm2, which should replace the conventional 50 high-power-field area. Standardized antigen retrieval is recommended in Japan but not in the NCCN and ESMO guidelines. Evidence of antigen retrieval during KIT immunostaining is lacking. Because GIST is a rare disease and diagnostic concordance among pathologists is not obtainable in some cases [17], consultation with or a second pathological examination by pathologists specializing in sarcoma is recommended in the ESMO guidelines.

Pathological diagnosis of gastrointestinal stromal tumor (GIST) by immunohistochemistry and genotyping. The algorithm for the pathological diagnosis of GIST is shown. The number sign means solitary fibrous tumors should be ruled out. DOG1 discovered on GIST-1, HE hematoxylin–eosin staining
Genotyping
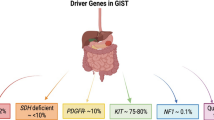
Mutation testing, at least for the KIT and PDGFRA genes, is recommended when TKIs, such as imatinib, sunitinib, and regorafenib, are to be used. KIT mutations (present in 80 % of primary GISTs) are commonest in exon 11 (65 %), followed by exon 9 (8 %), and are rarely found in exons 13 and 17. Most GISTs caused by KIT exon 11 or 13 mutations are naïve to imatinib. KIT exon 9 mutations are associated with a nongastric location, clinicopathologically aggressive features, and hyposensitivity to imatinib. GISTs with KIT exon 17 mutations are rare and some of them (e.g., D816V) are resistant to imatinib. PDGFRA mutations (present in 10 % of primary GISTs) are common in tumors of the stomach and have epithelioid features as well as indolent behaviors. The commonest mutation of PDGFRA, D842V, is associated with resistance to imatinib, sunitinib, and regorafenib. Approximately 10 % of GISTs are negative for KIT and PDGFRA mutations; these are referred to as wild-type GISTs. Wild-type GISTs are heterogeneous in genotype and may include mutations in HRAS, NRAS, BRAF, NF1 or the SDH complex (Table 1). Wild-type GISTs may be considered insensitive to imatinib. A mutation analysis may add prognostic information for GIST patients, especially for some specific subtypes, and genotyping can provide critical biomarkers to predict the activity of TKIs. Pathology reports may include, at least, the pathological diagnosis of GIST, tumor origin, presence of preoperative or intraoperative rupture, histological type, maximal size (cm), mitotic index (area of 5 mm2), surgical margin, immunohistochemical findings (KIT, DOG1, CD34, desmin, S100, Ki67), presence of pathological necrosis, risk stratification, and KIT and PDGFRA mutations.
There are several subsets of GISTs with features distinct from those of conventional KIT- or PDGFRA-mutated GISTs, including pediatric GIST, neurofibromatosis type 1 associated GIST (NF1-GIST), Carney–Stratakis syndrome, the Carney triad, and familial GISTs (Table 1):
-
1.
Pediatric GISTs, which are predominantly found in the female stomach, are frequently associated with predominant epithelioid features, lymph node metastasis, and mutations in the SDH complex. These tumors are sometimes multicentric and/or multinodular, and typically progress slowly. SDH-mutated GISTs are thought to be insensitive to imatinib, but sunitinib may work to some extent [10, 18].
-
2.
NF1–GISTs are marked by wild-type and multicentric tumors, are predominantly located in the small intestine, and are relatively indolent in terms of clinical and pathological features. NF1–GISTs are insensitive to imatinib.
-
3.
Carney–Stratakis syndrome is caused by germline loss-of-function mutations in SDH genes, including subunits A, B, C, and D, and is characterized by a dyad of gastric GIST and paraganglioma.
-
4.
The Carney triad is typically marked by gastric GISTs, paraganglioma, and pulmonary chondromas and may be accompanied by an epigenetic loss of SDH expression.
-
5.
Familial GISTs with germline mutations in either the KIT gene or the PDGFRA gene present as autosomal dominant traits and are associated with the presence of multiple GISTs in the gastrointestinal tract that are found in relatively young individuals [19].
Diagnostic imaging
Most GISTs are detected by endoscopy as a submucosal tumor (SMT), and the pathological diagnosis is often made after surgery. When small esophageal or gastric nodules (SMTs smaller than 2 cm) having no high-risk features are detected, they can usually be followed by periodic endoscopic ultrasonography (EUS) until the tumors increase in size or become symptomatic (Fig. 2), even if they are histologically GISTs [3, 4, 16]. Alternatively, the decision-making process can be shared with patients regarding whether to make a histological diagnosis—for example, by EUS-guided fine-needle aspiration (EUS-FNA) biopsy, or whether the patient should undergo further treatment. Although evidence to determine the optimal follow-up schedule is lacking, most guidelines recommend an initial short-term follow-up within 6 months by EUS (Fig. 2), followed by a more relaxed follow-up when there is no evidence of growth, high-risk features, or symptoms [3–5]. A recent retrospective study indicates that a relaxed follow-up did not worsen the prognosis of gastric GIST patients [12]. For rectal GISTs, however, the ESMO guidelines recommend surgical resection regardless of tumor size because the risk of rectal GIST is high and local control is critical. Although endoscopic removal of small GISTs has been reported, the safety and oncologic outcomes have not been established owing to the risks of positive margins, tumor spillage, and potential perforation. Therefore, endoscopic resection of SMTs is an investigational measure and should be performed only as part of a clinical trial in specialized centers [5, 6].

Diagnostic and therapeutic strategies for histologically undiagnosed gastric submucosal tumor (SMT) and histologically diagnosed gastric gastrointestinal stromal tumor (GIST). High-risk features include ulceration, irregular borders, internal heterogeneity, enlargement of regional lymph nodes, and an increase in size during follow-up. CT computed tomography, EUS endoscopic ultrasonography, EUS-FNA endoscopic-ultrasonography-guided fine-needle aspiration biopsy
EUS-FNA biopsy may provide the most reliable histological diagnosis of SMTs before surgery. Its indications include histologically undiagnosed SMTs that may require medical or surgical treatment depending on their histological characteristics, such as neoadjuvant therapy for marginally resectable GISTs. EUS-FNA biopsy is not recommended for tumors for which it has already been planned that they will be resected by surgery, undoubtedly benign tumors, and small tumors (less than 2 cm) [20]. Because of the diagnostic limitations of endoscopy for GISTs/SMTs showing extrinsic growth, contrast-enhanced computed tomography (CT) and/or EUS is also recommended for GISTs/SMTs larger than 2 cm in an initial workup to evaluate whole images of tumors and high-risk features [5, 12, 16], unless they are obviously benign tumors. The Japanese GIST guidelines recommend that GISTs/SMTs larger than 5 cm, except for definitely benign tumors, should be resected by surgery and should be subjected to subsequent pathological examinations [5, 16].
Prognostic factors and risk stratification
Independent prognostic factors for GIST include the mitotic index, tumor size, tumor location (gastric vs. nongastric), and tumor rupture [21]. Tumor rupture should be considered separately with regard to whether it occurred before or during surgery. Although the type of mutation(s) may add important prognostic information for risk assessment, the four factors mentioned above provide much more useful information in the prognostic stratification than the genotype does [22, 23].
Discrimination of benign GIST from malignant GIST by simply tumor diameter or mitotic index is not yet feasible; therefore, risk classification and nomograms have been introduced [23–26]. Of these, a risk-stratification procedure using tumor size and mitotic index, the National Institutes of Health (NIH) classification, is the method most frequently used in clinical trials because of the historical context [24], whereas the risk-classification method proposed by Miettinen and Lasota [25] that incorporates tumor size, number of mitoses, and tumor location is commonly used in daily clinical practice. The more recently proposed “modified NIH classification” is defined by four factors—number of mitoses, size, location, and rupture—and might offer advantages in the selection of patients who may require adjuvant therapy [23]. Nomograms can be used to estimate an individual’s risk of recurrence [26] and may be useful for individual decision-making with respect to adjuvant therapy. When tumor size and mitotic index are near the cut-off values, patients and physicians may discuss the information pertaining to estimated recurrence risk obtained from prognostic contour maps [23].
Surgery for primary GIST
Surgery remains the only modality that can offer a permanent cure of GIST, and complete surgical resection avoiding tumor rupture and injuries to the pseudocapsule is the initial treatment for primary and localized GISTs when the risk of morbidity and death from surgery is acceptable. The aims of surgery include complete resection with macroscopic and microscopic negative margins and functional preservation by wedge resection, when applicable. The management of a positive microscopic margin after macroscopic complete resection is not well defined, and options may include re-excision, watchful waiting, and postoperative imatinib therapy. The information regarding the margin status and postoperative therapy should be shared with patients, and a multidisciplinary team should be involved in clinical decision-making. A recent retrospective analysis of clinical studies suggested that the margin status may have no significant prognostic effect in this era of targeted therapy [27]. Lymph node metastasis is very rare in GIST, and prophylactic dissection of lymph nodes is not necessary, except for the SDH-mutated GISTs [9], for which pickup dissection of swollen lymph nodes may be indicated.
Laparoscopic surgery may be successful for small gastric GISTs under the same oncological principles as for open surgery. In laparoscopy, direct handling of tumors with forceps is contraindicated, and a plastic bag should be used to minimize the risk of tumor seeding when the tumor samples are removed. Several retrospective cohort studies have suggested that laparoscopic resection is feasible and safe for gastric GISTs smaller than 5 cm and is less invasive than open surgery, with similar oncological outcomes [28]. The ESMO guidelines state that a laparoscopic approach is acceptable for small GISTs; however, it is not recommended for large tumors because of the risk of tumor rupture. Likewise, the NCCN guidelines state that laparoscopic resection is a reasonably safe and feasible procedure for patients with gastric GISTs 5 cm or smaller and that data on laparoscopic resection of GISTs other than gastric ones or ones larger than 5 cm are limited and laparoscopic surgery for these GISTs is not always recommended. The indications for laparoscopic surgery may depend on the anatomic site, developmental ways of tumors (e.g., intraluminal or extraluminal growth), and possibly on the level of experience of the multidisciplinary team. The indications for and role of laparoscopic or laparoscopic-assisted surgery have not been determined for GISTs larger than 5 cm or intestinal GISTs.
Medical therapy for recurrent/metastatic GIST
Imatinib mesylate
Imatinib mesylate is a first-line standard therapy for inoperable, metastatic, or recurrent GISTs (Fig. 3). The standard dosage is 400 mg/day. The NCCN and ESMO guidelines recommend a higher dosage for KIT exon 9-mutated GISTs because the higher dosage (800 mg/day) showed a longer progression-free survival (PFS) in such cases in a clinical trial [29]. However, treatment with more than 400 mg/day is not reimbursed in some countries, including Japan. The Japanese guidelines indicate that a higher dosage for exon 9-mutated GISTs is an optional approach. Previous reports found that a higher dosage of imatinib was associated with severer toxic effects than the standard dosage [31, 32], and individual optimization of imatinib therapy is mandatory. Interruption of imatinib treatment is accompanied by disease progression [33], so imatinib therapy should be continued indefinitely when tolerable, even after a complete response or macroscopic resection of residual tumors. It has been reported that half to two thirds of patients with metastatic/recurrent GISTs may show an objective response after imatinib treatment, and the median PFS is more than 2 years, with nearly 15 % of patients showing a durable response lasting more than 10 years [30, 31, 33, 34]. It may take several months to obtain a therapeutic effect in some cases, and the median time to response was 3 months [2]. More importantly, patients with stable disease lasting more than 6 months show oncological outcomes similar to those with an objective response [34], suggesting that careful monitoring of the tumor response is important in the early phases of treatment. However, 10–15 % of GIST patients show intolerance or resistance (primary resistance) to imatinib.

Treatment algorithm for unresectable, metastatic, or recurrent gastrointestinal stromal tumor (GIST). Interventions include surgical resection, radiofrequency ablation, and transcatheter arterial embolization for patients with limited progression. Tyrosine kinase inhibitors (TKI) include imatinib, sunitinib, and regorafenib, BSC best supportive care, CR complete response, PD progressive disease, PR partial response, SD stable disease
Biomarkers of the activity of imatinib may include the genotype and plasma levels of the drug [35, 36]. GISTs with KIT exon 11 mutations are most sensitive to imatinib, whereas those with KIT exon 9 mutations are less sensitive and may require a higher dosage (800 mg/day) to achieve longer PFS. GISTs with specific mutations, such as PDGFRA exon 18 (D842V) or KIT exon 17 (D816V) mutations, are resistant to imatinib [37]. Wild-type GISTs, which have no mutation in the KIT and PDGFRA genes, are also thought to be insensitive to imatinib. Thus, mutation testing is recommended when imatinib is being considered for treatment. Suboptimal plasma levels of imatinib might be associated with short PFS [38]. Assessment of the plasma drug level may be useful when there are unexpected toxic effects, suspected poor adherence, possible major drug–drug interactions, or unexpected early progressive disease under standard dosing.
Resistance to imatinib may include primary and secondary resistance: the former is associated with progressive disease within 6 months and the latter is associated with progressive disease after 6 months. The major causes of primary resistance are genotypes such as the D842V mutation, wild-type subtypes, and some KIT exon 9 mutations; those associated with secondary resistance are new mutations in two kinase domains that occur in the ATP-binding pocket or activation loop during imatinib therapy [39]. Secondary mutations are more frequently observed in GISTs with KIT exon 11 mutations than in those with KIT exon 9 mutations. Secondary mutations show clonal evolution, and newly acquired mutations are highly heterogeneous with regard to the metastatic sites in a patient, and sometimes even within a particular tumor nodule, although the primary mutation is the same throughout all lesions.
Sunitinib malate
When GIST patients have progressive disease under imatinib treatment or are unable to tolerate imatinib because of adverse events, sunitinib malate (Sutent; Pfizer, New York, NY, USA) is recommended [40]. For some imatinib-resistant GISTs showing focal progression, resistant lesions may be treated by surgical resection, radiofrequency ablation, or transcatheter arterial embolization (Fig. 3). Surgery for limited progression has been shown to lead to a PFS of 6–12 months in retrospective studies [41–43], suggesting that these approaches for limited progression with continuing imatinib treatment may be useful. These treatments, however, are not well established and should be performed as investigational therapy by a multidisciplinary sarcoma team. The other option is dosage escalation of imatinib (to 800 mg/day), which may result in a prolongation of the median time-to-progression by 3 months [44].
Sunitinib is a multitarget inhibitor that inhibits KIT, platelet-derived growth factor receptors, vascular endothelial growth factor receptors 1–3, colony stimulating factor 1 receptor, and RET. The drug (50 mg/day) was initially approved for imatinib-resistant disease or intolerant patients with a 4-week-on/2-week-off schedule; the continuous use of 37.5 mg/day was later approved in the USA and EU but has not been approved in Japan. The dose and schedule may be individualized depending on patient response and adverse events. The reported response rate was nearly 10 %, and the clinical benefit rate was approximately 50 %, with a median PFS of 8 months, which was more than four times longer than that for the placebo [40]. The commonest treatment-related adverse events show a profile different from that for imatinib and are generally severer than those for imatinib. The activity of sunitinib is related to the primary and secondary mutations. With regard to the primary mutations, patients with KIT exon 9-mutated GISTs and wild-type GISTs receive more benefit from sunitinib treatment than do those with KIT exon 11 mutations. Regarding secondary mutations, patients who had GISTs with secondary mutations in the ATP-binding domain showed better responses and a better prognosis under sunitinib treatment than did those with mutations in the activation loop domain [45].
Regorafenib
Regorafenib, another multitarget inhibitor, inhibits KIT, platelet-derived growth factor receptors, vascular endothelial growth factor receptors 1-3, fibroblast growth factor receptor, RET and BRAF. Regorafenib (160 mg/day) was initially used for imatinib- and sunitinib-resistant GIST with a 3-week-on/1-week-off regimen and resulted in a response rate of 4.5 %, a clinical benefit rate of nearly 50 %, and a median PFS of 5 months [46]. Therefore, the third-line therapy for GISTs progressing under sunitinib treatment is regorafenib (Fig. 3). Another option is rechallenge with imatinib after progression under sunitinib treatment, which showed a twofold increase in PFS (1.8 months) compared with placebo [47]. Surgical treatment for focally progressing lesions under sunitinib treatment may work for exceptional cases [48].
Multidisciplinary management
After complete resection, nearly 60 % of GIST patients are cured by surgery alone; however, the other 40 % have relapses and require additional targeted therapy [21]. To improve the prognosis of patients with a substantial risk of recurrence, all the guidelines recommend adjuvant therapy with imatinib for 3 years, which improves not only relapse-free survival but also the overall survival of high-risk patients or those with ruptured GISTs [49]. Spontaneously ruptured GISTs, which may lead to spillage of tumor cells in the abdominal cavity, are thought to be accompanied by a very high risk of peritoneal recurrence. The optimal duration of adjuvant therapy for these patients is currently unknown. The use of adjuvant therapy is not recommended for very low risk or low-risk GIST, but there is no consensus for intermediate-risk GIST. The expected duration of treatment and the risks and benefits of treatment should be shared with patients. Mutation testing is critical in decision-making regarding the use of adjuvant therapy. None of the guidelines recommend adjuvant imatinib therapy for PDGFRA D842V mutations. In addition, wild-type GISTs are not considered candidates for adjuvant treatment; however, evidence is lacking for this contraindication. For patients with exon 9 mutations, a higher dosage of imatinib (800 mg/day) may be considered for adjuvant therapy, but there is a lack of evidence and some regulatory limitations, especially in Japan.
When patients are expected to have considerable morbidity and loss of organ functions after initial surgery, and when safety of surgery and organ-function sparing are anticipated after cytoreduction, preoperative imatinib therapy is recommended for very large and marginally resectable GISTs. Preoperative imatinib therapy does not increase the risk of complications of surgery, and the treatment has been shown to be feasible and safe; however, its long-term prognostic effects are still unclear [50]. Approximately 6 months of preoperative therapy may be considered if imatinib is active, but there is no established evidence regarding the length of preoperative therapy. In addition, these patients may require adjuvant therapy for 3 years to improve their prognosis. If the tumors progress during preoperative therapy, surgery is recommended after imatinib treatment has been promptly stopped. Thus, an initial evaluation of the activity of imatinib— for instance, within 1month of treatment starting—is important.
Monitoring and follow-up
Imaging follow-up after treatment
All the clinical practice guidelines contain a follow-up policy based on expert consensus. However, the recommendations for follow-up differ among the NCCN, ESMO, and Japanese and other countries’ guidelines in some aspects. A small tumor burden is associated with a better prognosis for TKI therapy. The objectives of follow-up after complete surgery may be early detection and treatment of relapses [51]. Abdominal and pelvic CT with contrast medium is sufficient for conventional follow-up of GIST patients because metastases outside the abdomen are very uncommon. Magnetic resonance imaging is an alternative to CT, especially in young patients. The frequency of imaging should be adjusted according to the risk of recurrence and the timing and conditions of treatment [52]. NIH, Armed Forces Institute of Pathology, or modified NIH risk stratification should be performed when the risk of recurrence is estimated after surgery. Annual abdominal CT for 5 years after surgery is thought to suffice for most patients with a less than intermediate risk of recurrence [51]. The trade-offs between early detection of recurrence and cumulative radiation exposure from repeated CT should be considered for patients with very low risk and low-risk GISTs. Recurrence risk after surgery is highest during the initial few years after surgery and decreases gradually thereafter, and the patients being treated with adjuvant imatinib therapy are at low risk when imatinib is active. However, their risk of recurrent GIST increases substantially during the first few years after the discontinuation of adjuvant imatinib therapy [49]. Thus, for high-risk GIST patients treated with adjuvant therapy, follow-up imaging may be done at 6-month intervals during the treatment, every 3–4 months during the first 2 years after adjuvant therapy has been stopped, and then once every 6–12 months for up to 10 years after surgery [51]. When patients have no adjuvant therapy, an interval of 3–4 months between imaging studies may be recommended during the initial few years after surgery.
Imaging in response evaluation
Evaluating the response is occasionally challenging, especially in the early and late phases of TKI therapy. The effects of imatinib may appear as tumor shrinkage and a decrease in CT density in the presence of contrast enhancement [53]. In principle, it is advisable that the response to TKIs be evaluated according to the Response Evaluation Criteria in Solid Tumors [54]. In some cases, however, the tumor size may increase in spite of a decrease in tumor density and substantial symptomatic improvement after imatinib therapy; this increase is associated with a subsequent gradual decrease in size and eventually durable stabilization of the disease. Thus, modified CT response evaluation criteria could be applicable for imatinib therapy [3, 53]. This is not always true for sunitinib and regorafenib [55]. Disease progression may present as new lesions, a significant increase (more than 10 %) in the size of existing tumors, or the appearance of small intratumoral nodules with contrast enhancement even if there is no change in the overall tumor size [56]. Although conclusive data are lacking regarding the optimal monitoring interval during imatinib therapy, follow-up with CT every 3–6 months seems reasonable. When progression is suspected, the imaging frequency should be increased, and magnetic resonance imaging or contrast-enhanced ultrasonography may be considered as an alternative evaluation measure. Both 2-deoxy-2-[18F]fluoro-d-glucose positron emission tomography and positron emission tomography–CT have proven to be highly sensitive in the early assessment of the tumor response and are thought to be useful in cases with confusing responses in CT or in the early prediction of the response (e.g., preoperative treatment). However, a small proportion of GISTs (10–20 %) have no [18F]fluorodeoxyglucose uptake, and this modality is not always reimbursed in response evaluation, especially in Japan.
Comments
The Japanese clinical practice guidelines for GIST were updated in 2014, and version 3.0 has been published in Japanese. This review is based on the ESMO guidelines, the new Japanese guidelines, and discussions with Asian experts. An official report by these experts will be published soon.
Toshirou Nishida, Seiichi Hirota, and Yuko Kitagawa are panel members for the Japanese clinical practice guidelines for GIST, Jean-Yves Blay is a panel member for the ESMO clinical practice guidelines for GIST, and Yoon-Koo Kang is a panel member for the clinical practice guidelines for GIST in Korea.
Abbreviations
- CT:
-
Computed tomography
- DOG1:
-
Discovered on gastrointestinal stromal tumor 1
- ESMO:
-
European Society for Medical Oncology
- EUS:
-
Endoscopic ultrasonography
- EUS-FNA:
-
Endoscopic-ultrasonography-guided fine-needle aspiration
- GIST:
-
Gastrointestinal stromal tumor
- NCCN:
-
National Comprehensive Cancer Network
- NF1-GIST:
-
Neurofibromatosis type 1 associated gastrointestinal stromal tumor
- NIH:
-
National Institutes of Health
- PFS:
-
Progression-free survival
- SDH:
-
Succinate dehydrogenase
- SDHB:
-
Succinate dehydrogenase iron–sulfur subunit (subunit B)
- SMT:
-
Submucosal tumor
- TKI:
-
Tyrosine kinase inhibitor
References
Hirota S, Isozaki K, Moriyama Y, Kanakura Y, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Tunio GM, Matsuzawa Y, Shinomura Y, Kitamura Y. Gain-of-function mutation of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80.
Demetri GD, von Mehren M, Antonescu CR, DeMatteo RP, Ganjoo KN, Maki RG, Pisters PW, Raut CP, Riedel RF, Schuetze S, Sundar HM, Trent JC, Wayne JD. NCCN Task Force report: update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Netw. 2010;8(Suppl 2):S1–41.
ESMO/European Sarcoma Network Working Group. Gastrointestinal stromal tumours: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii21–6.
Nishida T, Hirota S, Yanagisawa A, Sugino Y, Minami M, Yamamura Y, Otani Y, Shimada Y, Takahashi F, Kubota T. Clinical practice guideline for gastrointestinal stromal tumor (GIST) in Japan. Int J Clin Oncol. 2008;13:416–30.
Kang YK, Kang HJ, Kim KM, Sohn T, Choi D, Ryu MH, Kim WH, Yang HK, Korean GIST Study Group (KGSG). Clinical practice guideline for accurate diagnosis and effective treatment of gastrointestinal stromal tumor in Korea. Cancer Res Treat. 2012;44:85–96.
Yeh CN, Hwang TL, Huang CS, Lee PH, Wu CW, Chen-Guo K, Jan YY, Chen MF, Taiwan Surgical Society of Gastroenterology. Clinical practice guidelines for patients with gastrointestinal stromal tumor in Taiwan. World J Surg Oncol. 2012;10:246.
Nilsson B, Bümming P, Meis-Kindblom JM, Odén A, Dortok A, Gustavsson B, Sablinska K, Kindblom LG. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era—a population-based study in western Sweden. Cancer. 2005;103:821–9.
Ma GL, Murphy JD, Martinez ME, Sicklick JK. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298–302.
Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35:1712–21.
Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet. 2013;382:973–83.
Park CH, Kim EH, Jung DH, Chung H, Park JC, Shin SK, Lee YC, Kim H, Lee SK. Impact of periodic endoscopy on incidentally diagnosed gastric gastrointestinal stromal tumors: findings in surgically resected and confirmed lesions. Ann Surg Oncol. 2015. doi:10.1245/s10434-015-4517-0.
Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, Hosoya Y, Nakajima T, Funata N. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37:1527–35.
Agaimy A, Wünsch PH, Dirnhofer S, Bihl MP, Terracciano LM, Tornillo L. Microscopic gastrointestinal stromal tumors in esophageal and intestinal surgical resection specimens: a clinicopathologic, immunohistochemical, and molecular study of 19 lesions. Am J Surg Pathol. 2008;32:867–73.
Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002;160:1567–72.
Nishida T, Kawai N, Yamaguchi S, Nishida Y. Submucosal tumors: a comprehensive guide for the diagnosis and therapy of gastrointestinal submucosal tumors. Dig Endosc. 2013;25:479–89.
Ray-Coquard I, Montesco MC, Coindre JM, Dei Tos AP, Lurkin A, Ranchère-Vince D, Vecchiato A, Decouvelaere AV, Mathoulin-Pélissier S, Albert S, Cousin P, Cellier D, Toffolatti L, Rossi CR, Blay JY. Sarcoma: concordance between initial diagnosis and centralized expert review in a population-based study within three European regions. Ann Oncol. 2012;23:2442–9.
Maki R, Blay J-Y, Demetri GD, Fletcher JA, Joensuu H, Martín-Broto J, Nishida T, Reichardt P, Schöffski P, Trent JC. A brief history of GIST: redefining the management of solid tumors. Oncologist. 2015;20:823–30.
Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y. Familial gastrointestinal stromal tumors with germ line mutation of the KIT gene. Nat Genet. 1998;19:323–4.
Dumonceau JM, Polkowski M, Larghi A, Vilmann P, Giovannini M, Frossard JL, Heresbach D, Pujol B, Fernández-Esparrach G, Vazquez-Sequeiros E, Ginès A, Indications, results, and clinical impact of endoscopic ultrasound (EUS)-guided sampling in gastroenterology: European Society of Gastrointestinal Endoscopy (ESGE) clinical guideline. Endoscopy. 2011;43:897–912.
Joensuu H, Vehtari A, Riihimäki J, Nishida T, Steigen SE, Brabec P, Plank L, Nilsson B, Cirilli C, Braconi C, Bordoni A, Magnusson MK, Linke Z, Sufliarsky J, Massimo F, Jonasson JG, Dei Tos AP, Rutkowski P. Risk of gastrointestinal stromal tumour recurrence after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265–74.
Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, Blackstein ME, Blanke CD, Demetri GD, Heinrich MC, von Mehren M, Patel S, McCarter MD, Owzar K, DeMatteo RP. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563–70.
Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, Nilsson B, Braconi C, Bordoni A, Magnusson MK, Sufliarsky J, Federico M, Jonasson JG, Hostein I, Bringuier P-P, Emile J-F. KIT and PDGFRA mutations and the risk of gastrointestinal stromal tumor recurrence. J Clin Oncol. 2015;33:634–42.
Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, Miettinen M, O’Leary TJ, Remotti H, Rubin BP, Shmookler B, Sobin LH, Weiss SW. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol. 2002;33:459–65.
Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70–83.
Gold JS, Gönen M, Gutiérrez A, Broto JM, García-del-Muro X, Smyrk TC, Maki RG, Singer S, Brennan MF, Antonescu CR, Donohue JH, DeMatteo RP. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045–52.
McCarter MD, Antonescu CR, Ballman KV, Maki RG, Pisters PW, Demetri GD, Blanke CD, von Mehren M, Brennan MF, McCall L, Ota DM, DeMatteo RP, American College of Surgeons Oncology Group (ACOSOG) Intergroup Adjuvant Gist Study Team. Microscopically positive margins for primary gastrointestinal stromal tumors: analysis of risk factors and tumor recurrence. J Am Coll Surg. 2012;215:53–9.
Chen K, Zhou YC, Mou YP, Xu XW, Jin WW, Ajoodhea H. Systematic review and meta-analysis of safety and efficacy of laparoscopic resection for gastrointestinal stromal tumors of the stomach. Surg Endosc. 2015;29:355–67.
Gastrointestinal Stromal Tumor Meta-Analysis Group. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1640 patients. J Clin Oncol. 2010;28:1247–53.
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–34.
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–32.
Blay JY, Le Cesne A, Ray-Coquard I, Bui B, Duffaud F, Delbaldo C, Adenis A, Viens P, Rios M, Bompas E, Cupissol D, Guillemet C, Kerbrat P, Fayette J, Chabaud S, Berthaud P, Perol D. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol. 2007;25:1107–13.
Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, Wehre E, Nikolova Z, Joensuu H. Long-term results from a randomized phase II trial of standard—versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5.
Le Cesne A, Van Glabbeke M, Verweij J, Casali PG, Findlay M, Reichardt P, Issels R, Judson I, Schoffski P, Leyvraz S, Bui B, Hogendoorn PC, Sciot R, Blay JY. Absence of progression as assessed by response evaluation criteria in solid tumors predicts survival in advanced GI stromal tumors treated with imatinib mesylate: the intergroup EORTC-ISG-AGITG phase III trial. J Clin Oncol. 2009;27:3969–74.
Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDG-FRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–95.
Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–7.
Cassier PA, Fumagalli E, Rutkowski P, Schöffski P, Van Glabbeke M, Debiec-Rychter M, Emile JF, Duffaud F, Martin-Broto J, Landi B, Adenis A, Bertucci F, Bompas E, Bouché O, Leyvraz S, Judson I, Verweij J, Casali P, Blay JY, Hohenberger P. Outcome of patients with platelet-derived growth factor receptor alpha–mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. 2012;18:4458–64.
Demetri GD, Wang Y, Wehrle E, Racine A, Nikolova Z, Blanke CD, Joensuu H, von Mehren M. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141–7.
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M, Fletcher CD, Sandau K, McDougall K, Ou WB, Chen CJ, Fletcher JA. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74.
Demetri GD, van Oosterom AT, Garrett CR, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38.
Raut CP, Posner M, Desai J, Morgan JA, George S, Zahrieh D, Fletcher CD, Demetri GD, Bertagnolli MM. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol. 2006;24:2325–31.
DeMatteo RP, Maki RG, Singer S, Gonen M, Brennan MF, Antonescu CR. Results of tyrosine kinase inhibitor therapy followed by surgical resection for metastatic gastrointestinal stromal tumor (GIST). Ann Surg. 2007;245:347–52.
Hasegawa J, Kanda T, Hirota S, Fukuda M, Nishitani A, Takahashi T, Kurosaki I, Tsutsui S, Hatakeyama K, Nishida T. Surgical interventions for focal progression of advanced gastrointestinal stromal tumors under imatinib therapy. Inter J Clin Oncol. 2007;12:212–7.
Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, Schlemmer M, Van Glabbeke M, Brown M, Judson IR. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer. 2005;41:1751–7.
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, Cohen DP, Baum CM, Demetri GD. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–9.
Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, Badalamenti G, Blackstein M, Le Cesne A, Schöffski P, Maki RG, Bauer S, Nguyen BB, Xu J, Nishida T, Chung J, Kappeler C, Kuss I, Laurent D, Casali PG. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302.
Kang YK, Ryu MH, Yoo C, Ryoo BY, Kim HJ, Lee JJ, Nam BH, Ramaiya N, Jagannathan J, Demetri GD. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2013;14:1175–82.
Raut CP, Wang Q, Manola J, Morgan JA, George S, Wagner AJ, Butrynski JE, Fletcher CD, Demetri GD, Bertagnolli MM. Cytoreductive surgery in patients with metastatic gastrointestinal stromal tumor treated with sunitinib malate. Ann Surg Oncol. 2010;17:407–15.
Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, Ramadori G, Hohenberger P, Duyster J, Al-Batran SE, Schlemmer M, Bauer S, Wardelmann E, Sarlomo-Rikala M, Nilsson B, Sihto H, Monge OR, Bono P, Kallio R, Vehtari A, Leinonen M, Alvegård T, Reichardt P. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265–72.
Wang D, Zhang Q, Blanke CD, Demetri GD, Heinrich MC, Watson JC, Hoffman JP, Okuno S, Kane JM, von Mehren M, Eisenberg BL. Phase II trial of neoadjuvant/adjuvant imatinib mesylate for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumors: long-term follow-up results of Radiation Therapy Oncology Group 0132. Ann Surg Oncol. 2012;19:1074–80.
Joensuu H, Martin-Brotob J, Nishida T, Reichardt P, Schöffski P, Maki RG. Follow-up strategies for patients with gastrointestinal stromal tumor treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611–7.
Joensuu H, Reichardt P, Eriksson M, Sundby Hall K, Vehtari A. Gastrointestinal stromal tumor: a method for optimizing the timing of CT scans in the follow-up of cancer patients. Radiology. 2014;271:96–103.
Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, Chen LL, Podoloff DA, Benjamin RS. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–9.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47.
Shinagare AB, Jagannathan JP, Kurra V, Urban T, Manola J, Choy E, Demetri GD, George S, Ramaiya NH. Comparison of performance of various tumour response criteria in assessment of regorafenib activity in advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib. Eur J Cancer. 2014;50:981–6.
Shankar S, vanSonnenberg E, Desai J, Dipiro PJ, Van Den Abbeele A, Demetri GD. Gastrointestinal stromal tumor: new nodule-within-a-mass pattern of recurrence after partial response to imatinib mesylate. Radiology. 2005;235:892–8.
Acknowledgments
This work was supported in part by a Grant-in-Aid from the Ministry of Health, Labor and Welfare, a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology, the National Cancer Center Research and Development Fund, and a grant from Yasuda Medical Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
T. Nishida has received funding for basic research from Novartis and Bayer, and honoraria for speeches from Bayer and Pfizer. J.-Y. Blay has received research support and honoraria from Novartis, Pfizer, and Bayer. Y.-K. Kang has received grants for research and honoraria for speeches from Novartis and Bayer. S. Hirota and Y. Kitagawa declare that they have no conflict of interest.
Ethical standards
All authors declare the trust and integrity of this review by their following the rules of good scientific practice, including the following: The manuscript has not been submitted to another journal simultaneously. The manuscript has not been published previously. A single study is not split into several parts to increase the quantity of submissions. No data have been fabricated or manipulated to support the conclusions. No data, text, or theories by others are presented as if they were those of the authors. Proper acknowledgments of other work have been given, quotation marks are used for verbatim copying of material, and permission has been secured for material that is copyrighted, if necessary. Consent to submit the article was received explicitly from all authors, as well as from the responsible authorities—tacitly or explicitly—at the institute/organization where the work was performed, before the work was submitted. All authors contributed sufficiently to the scientific work and therefore share collective responsibility and accountability for the descriptions.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nishida, T., Blay, JY., Hirota, S. et al. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer 19, 3–14 (2016). https://doi.org/10.1007/s10120-015-0526-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-015-0526-8