
Abstract
Two multicentre external quality assessments (EQA) for the molecular detection and genotyping of meticillin-resistant Staphylococcus aureus (MRSA) were arranged. Firstly, 11 samples containing various amounts of inactivated MRSA strains, meticillin-susceptible S. aureus (MSSA), meticillin-resistant coagulase-negative staphylococci (MRCoNS) or Escherichia coli were distributed to 82 laboratories. Samples containing 102 or 103 MRSA cells were correctly scored in only 16 and 46% of the datasets returned, respectively. Two of the used MSSA strains contained an SCCmec cassette lacking the mecA gene. There was a marked difference in the percentage of correct results for these two MSSA strains (37 and 39%) compared to the MSSA strain lacking the SCCmec cassette (88%). Secondly, a panel for MRSA genotyping, consisting of ten samples (two identical, three genetically related and five unique strains) was distributed to 19 laboratories. Seventy-three percent of the datasets recorded all samples correctly. Most pulsed-field gel electrophoresis (PFGE) protocols proved to be suboptimal, resulting in inferior resolution in the higher or lower fragment regions. The performance of molecular diagnostics for MRSA shows no significant changes since our first EQA in 2006. The first molecular typing results are encouraging. Both assessments indicate that programme expansion is required and that major performance discrepancies continue to exist.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adequate infection control of meticillin-resistant Staphylococcus aureus (MRSA) strongly depends on the speed and quality of (molecular) identification and characterisation strategies used by the clinical microbiological laboratory [1, 2].
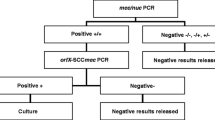
Over the past 4–5 decades, cultivation assays have been primarily used for the detection and subsequent identification of MRSA. However, cultivation requires prolonged incubation periods and, in general, clinically relevant meticillin resistance still needs to be confirmed by the detection of the mecA gene or its product. Nucleic acid amplification techniques (NAATs) offer benefits over traditional culture-based assays, in particular, a reduced time to identification and an improved specificity and sensitivity. Over the past decade, a range of commercial and in-house developed NAATs has been introduced. The sensitivity and specificity of these assays may be compromised, as a result of inhibition or false-positivity due to the presence of meticillin-resistant coagulase negative staphylococci (MRCoNS) or variability within the mec-resistance cassette. This may lead to false-negative results (new staphylococcal cassette chromosome mec [SCCmec] variants) or false-positive results (deletion of the mecA gene) [3–6].
After MRSA detection, genetic typing may be necessary in order to assess whether local cross-infection occurs and whether preventive measurements are mandatory. Currently, many different genotyping methods are in use in the diagnostic laboratory, but pulsed-field gel electrophoresis (PFGE) of SmaI digested genomic DNA still remains the most frequently used method [7]. Only when outbreaks are properly defined, adequate infection control measurements can be implemented.
The current multicentre external quality assessment (EQA) study determined the performance of molecular assays to detect MRSA and genotyping techniques to differentiate MRSA strains. The studies were coordinated by Quality Control for Molecular Diagnostics (QCMD) in Glasgow, Scotland.
Material and methods
EQA for molecular MRSA detection and identification
In August 2009, the EQA MRSA panel for MRSA detection and identification was distributed to 80 participating laboratories in 15 countries, along with detailed sample processing instructions. Participants were given 6 weeks to examine the samples and to report their results to the QCMD by using an online data collection system. The QCMD MRSA panel consisted of six samples containing 106, 105 (n = 2), 104, 103 and 102 CFU/ml MRSA bacterial cells, one meticillin-susceptible S. aureus (MSSA) sample, one sample containing MRCoNS, two samples containing MSSA harbouring an SCCmec cassette lacking the mecA gene and one sample containing Escherichia coli (Table 1). The production laboratory quantified the contents of the samples on the basis of colony counting, optical density measurements and real-time molecular amplification results. All bacterial samples were heat-inactivated for 10 min at 100°C.
EQA for MRSA genotyping
The EQA panel for MRSA genotyping was distributed to 19 participants in eight countries in August 2009. The panel consisted of ten samples of viable MRSA strains in Müller Hinton broth. Genetic relatedness of the MRSA panel was originally determined with PFGE [8]. The current panel consisted of two identical strains, three genetically related strains and five unique strains (Table 2). Genotype and subtype were reported by the production laboratory. A different letter signifies the detection of a different genotype, whereas a different number signifies the detection of a different subtype. All data were reported in relation to the reference strain in panel sample MRSATP09-01.
The QCMD Neutral Office analysed the data, which was anonymously released to all participants in a detailed EQA final report.
Results
EQA for molecular MRSA detection and identification
Out of the 80 participants, 68 (85%) responded. Twelve participants did not return results. Five of these withdrew officially, indicating ‘assay not offered’ (n = 2), ‘internal issues’ (n = 1) and ‘other’ (n = 2) as the reason for withdrawal.
The following commercial amplification assays were used for MRSA detection: BAG HealthCare Hyplex StaphyloResist (n = 1) (BAG Healthcare, Lich, Germany), BD Diagnostic GeneOhm MRSA Assay (n = 13) (BD Diagnostics, GeneOhm, San Diego, California), BD Diagnostics GeneOhm Staph SR Assay (n = 5) (BD Diagnostics , GeneOhm), Cepheid IDI MRSA (n = 1) (Cepheid, Sunnyvale, California), Cepheid Xpert MRSA Test (n = 11) (Cepheid), Cepheid Xpert MRSA/SA Test (n = 2) (Cepheid), Roche LightCycler MRSA Advanced Test (n = 4) (Roche Diagnostics, Basel, Switzerland), TIB MOLBIOL LightMix Kit MRSA (n = 2) (TIB MolBiol, Berlin, Germany) and Hain Lifescience GenoQuick MRSA (n = 2) (Hain Lifescience, Nehren, Germany). This diversity overlaps with the spectrum of currently available commercial tests.
All results are summarised in Table 3. Results for the panel samples with 106 MRSA cells (MRSA09-05), 105 MRSA cells (MRSA09-04 and MRSA09-08) and 104 MRSA cells (MRSA09-07) were reported correctly in 100, 100, 99 and 98% of the datasets, respectively. The samples containing lower amounts, MRSA09-10 (103 CFU/ml) and MRSA09-06 (102 CFU/ml), were reported correctly in only 46 and 16% of the datasets, respectively. No statistically significant differences in sensitivity or specificity could be seen between the different tests or between commercial and in-house testing. MRCoNS sample MRSA09-01 was correctly reported as MRSA-negative by 87% (40 out of 46) of the commercial polymerase chain reaction (PCR) tests and in 70% (23 out of 33) of the in-house PCR assays. MSSA sample MRSA09-03 was correctly reported as MRSA-negative by 89% (41 out of 46) of commercial PCR tests and in 85% (28 out of 33) of datasets generated by in-house PCR assays, respectively. The MSSA samples containing the SCCmec cassette but lacking the mecA gene (MRSA09-02 and MRSA09-09) were both incorrectly reported as positive by commercial PCR tests in 87% (40 out of 46). For in-house assays, these samples were reported incorrectly in 24% (8 out of 33) and 30% (10 out of 33), respectively. These percentages of incorrect results underline the need for improved specificity of these MRSA tests and, therefore, positive results should always be confirmed by a culture method or a second molecular test. For laboratories with high false-positivity rates or in regions with a low prevalence of MRSA, confirmation is essential [9]. For the E. coli sample, commercial PCR results were reported correctly in 87% (39 out of 45), whereas in-house PCR tests recorded correct results in 97% (29 out of 30).
EQA for MRSA genotyping
Out of the 19 potential participants, 14 (74%) responded. Four of the non-responders withdrew officially, indicating ‘panel used for research’ (n = 1) and ‘assay not offered’ (n = 3). The majority of datasets were generated by PFGE (n = 11), with the remainder generated by AFLP (n = 2) and spa typing (n = 2). Only eight participants (73%) scored all samples correctly, all with PFGE (Table 2).
Discussion
To maintain high-quality clinical care, quality control of molecular diagnostics is very important. The primary aim of our EQA programmes was to assess the proficiency of laboratories in the molecular detection and characterisation of MRSA strains. Here, we conclude that the molecular detection of MRSA in samples with high CFU counts is reliable, which can, and has been, implemented in various laboratory settings with confidence. All tests performed equally well. However, for direct molecular diagnostics, we have to conclude that current commercial and in-house tests do not meet the required quality criteria. The sensitivity of many tests is relatively low and the specificity needs to be improved. The FDA submission in 2004 of the IDI-MRSA test had a detection limit of 325 CFU/swab [10]. This is much more sensitive than documented in this study. Pre-enrichment of clinical samples leads to concentrations of MRSA exceeding 109 CFU/ml, which is higher than the concentrations of MRSA likely to be found in a patient sample and also those in this EQA panel. However, pre-enrichment reduces one of the major advantages offered by NAATs, namely, rapidity. In this year’s panel, only inactivated cells were present. As a consequence, pre-enrichment was not possible. This may have influenced the results of some laboratories. For the years to come, viable cells will be distributed, which is more similar to the real clinical situation.
This is the fourth year that the QCMD has offered the MRSA DNA EQA Programme and the number of participants has increased from 51 in 2006, 61 in 2007 and 74 in 2008 to 80 in 2009 [11]. Over the years, we observe a statistically non-significant decrease in the overall test sensitivity. The most pronounced discrepancies were observed in the low-concentration panel samples (103 and 102 CFU/ml). Conversely, the percentage of correct results showed an overall improvement for the ‘specificity’ samples (containing MSSA and MRCoNS or E. coli) and the ‘true-negative’ samples (Table 4). Again, this was not statistically significant. Still, these incorrect results underline the need for improved specificity of molecular MRSA tests and, therefore, positive results should always be confirmed by a culture method or a second molecular test. For laboratories with high false-positivity rates or in regions with a low prevalence of MRSA, confirmation is necessary.
In 2009, two MSSA samples harbouring an SCCmec cassette, but lacking the mecA gene, were included. There was a marked difference in the percentage of correct qualitative results for the MSSA strain containing the mecA gene compared to the two strains lacking it. These data show that confirmation on results generated using assays that only target the SCCmec cassette is mandatory.
In conclusion, the quality of molecular diagnostic tests still needs improvement, and proper and regular quality control and international standardisation for MRSA diagnostics should be mandatory for the years to come.
We present the first QCMD EQA programme for the genetic characterisation of MRSA strains. Clear differences in resolution were observed between the datasets. Some PFGE protocols, which were implemented by most of the participating laboratories, proved to be suboptimal, as a low level of discrimination in the high and/or low molecular weight fragments was observed in the majority of the results reported. This suggests the need for optimisation of the PFGE programme. This lack of resolution was most evident within the group of closely related MRSA strains in the panel (MRSATP09-08, MRSATP09-09 and MRSA09-10). These strains were incorrectly reported in 27% of datasets. Participants reported using a range of criteria for determining genotype and subtype. The guidance according to Tenover et al. was the most prominent method [12].
To improve the performance and quality of molecular diagnostics, both laboratories and manufacturers should be encouraged to participate in EQAs. The availability of EQA panels for detection and typing should also be developed for other important (nosocomial) infectious agents, including vancomycin-resistant enterococci (VRE) and extended-spectrum β-lactamase (ESBL)-producing enterobacteriaceae.
References
Brown DF, Edwards DI, Hawkey PM, Morrison D, Ridgway GL, Towner KJ, Wren MW (2005) Guidelines for the laboratory diagnosis and susceptibility testing of methicillin-resistant Staphylococcus aureus (MRSA). J Antimicrob Chemother 56(6):1000–1018
Weller TM (2000) Methicillin-resistant Staphylococcus aureus typing methods: which should be the international standard? J Hosp Infect 44(3):160–172
Ibrahem S, Salmenlinna S, Virolainen A, Kerttula AM, Lyytikäinen O, Jägerroos H, Broas M, Vuopio-Varkila J (2009) Carriage of methicillin-resistant Staphylococci and their SCCmec types in a long-term-care facility. J Clin Microbiol 47(1):32–37
Bartels MD, Boye K, Rohde SM, Larsen AR, Torfs H, Bouchy P, Skov R, Westh H (2009) A common variant of staphylococcal cassette chromosome mec type IVa in isolates from Copenhagen, Denmark, is not detected by the BD GeneOhm methicillin-resistant Staphylococcus aureus assay. J Clin Microbiol 47(5):1524–1527
Jansen WT, Beitsma MM, Koeman CJ, van Wamel WJ, Verhoef J, Fluit AC (2006) Novel mobile variants of staphylococcal cassette chromosome mec in Staphylococcus aureus. Antimicrob Agents Chemother 50(6):2072–2078
Ender M, Berger-Bächi B, McCallum N (2007) Variability in SCCmecN1 spreading among injection drug users in Zurich, Switzerland. BMC Microbiol 7:62
Ichiyama S, Ohta M, Shimokata K, Kato N, Takeuchi J (1991) Genomic DNA fingerprinting by pulsed-field gel electrophoresis as an epidemiological marker for study of nosocomial infections caused by methicillin-resistant Staphylococcus aureus. J Clin Microbiol 29(12):2690–2695
van Belkum A, van Leeuwen W, Kaufmann ME, Cookson B, Forey F, Etienne J, Goering R, Tenover F, Steward C, O'Brien F, Grubb W, Tassios P, Legakis N, Morvan A, El Solh N, de Ryck R, Struelens M, Salmenlinna S, Vuopio-Varkila J, Kooistra M, Talens A, Witte W, Verbrugh H (1998) Assessment of resolution and intercenter reproducibility of results of genotyping Staphylococcus aureus by pulsed-field gel electrophoresis of SmaI macrorestriction fragments: a multicenter study. J Clin Microbiol 36(6):1653–1659
Kerremans JJ, Maaskant J, Verbrugh HA, van Leeuwen WB, Vos MC (2008) Detection of methicillin-resistant Staphylococcus aureus in a low-prevalence setting by polymerase chain reaction with a selective enrichment broth. Diagn Microbiol Infect Dis 61(4):396–401
Huletsky A, Giroux R, Rossbach V, Gagnon M, Vaillancourt M, Bernier M, Gagnon F, Truchon K, Bastien M, Picard FJ, van Belkum A, Ouellette M, Roy PH, Bergeron MG (2004) New real-time PCR assay for rapid detection of methicillin-resistant Staphylococcus aureus directly from specimens containing a mixture of staphylococci. J Clin Microbiol 42(5):1875–1884
van Belkum A, Niesters HG, MacKay WG, van Leeuwen WB (2007) Quality control of direct molecular diagnostics for methicillin-resistant Staphylococcus aureus. J Clin Microbiol 45(8):2698–2700
Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B (1995) Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33(9):2233–2239
Acknowledgements
We acknowledge Deborah Kreft for her technical assistance in the preparation of the panels.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
te Witt, R., van Belkum, A., MacKay, W.G. et al. External quality assessment of the molecular diagnostics and genotyping of meticillin-resistant Staphylococcus aureus . Eur J Clin Microbiol Infect Dis 29, 295–300 (2010). https://doi.org/10.1007/s10096-009-0856-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-009-0856-8