
Abstract
There is mounting evidence indicating that the synovial fibroblasts (SFs) contribute to the pathogenesis of rheumatoid arthritis (RA). The present study showed the differential proteins expression pattern of SFs from patients with RA or osteoarthritis (OA) and healthy control. Cellular proteins of cultured SFs were subjected to 2-DE and visualized by silver nitrate staining. A total of 49 spots that were statistically and differentially overexpressed in RA or OA in comparison to healthy ones were identified by MALDI-TOF-MS, and 25 proteins were successfully identified. Western blot was used to further verify some of the differential proteins. These proteins included enzymatic and structural proteins, signal transduction proteins, calcium binding protein, etc. From all of the identified proteins, a number of proteins have been implicated that involved in the healthy or pathological SFs function (e.g., S100A4, S100A10, cathepsin D) or that have potential diagnostic and prognostic value for RA (α-enolase and TPI) or that may be the new therapeutic targets (Annexin, SOD, PRX).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To date, the complex molecular mechanisms of rheumatoid arthritis (RA) are not fully understood. Multiple cell types contribute to the pathogenic picture in rheumatoid synovium (pannus), and accumulating evidence suggests that synovial fibroblasts (SFs, also called fibroblast-like synovial cells) as part of a complex cellular network play an important role in the pathogenesis of rheumatoid arthritis.
In recent years, SFs have been studied intensively, and significant progress has been made in elucidating the specific features of these fibroblasts. First, SFs respond to, and themselves produce, inflammatory mediators; thus, they are important drivers of RA inflammation [1]. Second, several important proangiogenic cytokines and growth factors including fibroblast growth factor, vascular endothelial growth factor, and IL-18 are released by SFs. It suggests that SFs also contribute to blood vessel growth, which is necessary to sustain pannus formation and the arthritis process [2]. Third, direct contact between SFs and T cells leads to T-cell activation, inducing production of T-cell cytokines as well as T-cell proliferation, which suggests that SFs may play a direct role in the propagation, not only of inflammation but also of autoimmunity [3]. Fourth, moreover, it has been understood that although macrophage- and lymphocyte-secreted factors contributing to their activation, SFs do not merely respond to stimulation by proinflammatory cytokines but show a complex pattern of molecular changes also maintained in the absence of external stimulation [4].
Osteoarthritis (OA) is a chronic, degenerative disorder characterized by gradual loss of articular cartilage. It is generally believed to be a disease primarily caused by biomechanical alteration. It is clear now that profound inflammation appears during the course of the disease in many patients and that inflammatory infiltration can be found in the synovial membrane in OA joints [5]. That means that a cell-mediated specific immune response might be involved in the OA.
Although studying the characteristics and behavior of SFs in vitro has generated much of the understanding of the phenotypic changes they undergo in RA, the pathogenesis of SFs in RA remains uncertain. Previous work tried to analyze the protein expression pattern of SFs derived from the synovia of RA patients using two-dimensional gel electrophoresis (2-DE) and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) and identified 254 unique proteins, which likely implicated in the normal or pathological FLS function [6]. However, the protein expression pattern is not able to select the possible specific or significant proteins, although these specific or significant proteins might be presented in the pattern. Therefore, it is necessary to further study the molecular basis that contributes to inflammatory events. Technologies of differential display proteomics provide a platform for systematic comparative research at the protein level. It has been proven to be very valuable in the screening and identification of function-associated molecules, biomarkers, and drug targets. In our study, the differential proteomics analysis of SFs from RA, OA, and health control was applied to further explore the molecular basis of SFs function alteration.
Materials and methods
Isolation and culture of SFs
For 2-DE, synovial biopsy samples were obtained from 11 patients undergoing total knee replacement and seven donors of healthy individuals (surgical trauma). Five patients with RA fulfilling the American College of Rheumatology (ACR) criteria [7] and six patients with OA fulfilling the ACR criteria [8] were included in this study. All samples were obtained according to the guidelines approved by the Ethics Committee of Chongqing Southwest Hospital. SFs were isolated. Briefly, synovial tissue was digested with trypsin (0.5%) for 2 h at 37°C. Cells in the supernatant were collected and washed with phosphate-buffered saline by centrifugation at 800×g for 10 min. The cell pellet were resuspended with DMEM medium containing 10% fetal bovine serum and cultured at 37°C in a humidified 5% CO2 environment overnight. The nonadherent cells were discarded, while the adherent cells were cultured with growth medium. Once the cells were subconfluent at 80%, they were trypsinized and subcultured. Cells of the third passages were used in the further experiment.
Sample preparation
The cell pellet was dissolved in a sample buffer containing 7 M urea, 2 M thiourea, 4% CHAPS, 0.3% (w/v) Biolyte ampholytes (pH 3–10) and 75 mM dithiothreitol, followed by sonication at 10 μmol for 3 × 10 s. The samples were kept in a shaker for at least 4 h. The samples from either RA, or OA, or control were mixed respectively, and the proteins were quantified by a modified RC protein assay kit (Bio-Rad).
Two-dimensional electrophoresis
Four hundred micrograms of proteins were subjected to 2-DE. 2-DE was performed following the manufacturer’s instructions. After in-gel rehydration, immobilized pH gradient strips (17 cm, pH 3–10, NL; pH 4–7, respectively, Bio-Rad) were iso-electrically focused on the Protein IEF cell (Bio-Rad) at 20°C. Then, the strips were equilibrated. Finally, the strips were fixed with 0.5% agarose on top of 12% sodium dodecyl sulfate-polyacrylamide gel electrophoreses, and run in sets of two gels in the vertical Protein II Xi Cell (Bio-Rad) at 10 mA/gel for 30 min and 25 mA/gel. Gels were stained with the Silver Stain Plus Kit (Bio-Rad) following the manufacturer’s instructions. Each sample was run on duplicate gels.
Image acquisition and data analysis
The silver-stained gels were scanned using a GS800 Calibrated Densitometer (Bio-Rad). Digitized images were analyzed with the PDQuest v.7.3.1 computer software (Bio-Rad). The spots were detected by using the spot detection wizard, and the background was removed by applying the floater background subtraction. Proteomic differences in each gel were analyzed by compared with the matched set standard image containing the match information of all the gels created by the software itself.
Protein identification by MALDI-TOF-MS
The significant different spots in each gel were manually excised, dehydrated with acetonitrile. The proteins were reduced and alkylated. Finally, the gel pieces were thoroughly washed and were digested in 5 μl of modified trypsin solution (1 ng/μl in 25 mM ammonium bicarbonate). The digestion reaction was stopped by 1 μl of 5% TFA.
The digestions were dropped onto the AnchorChipTM (Bruker Daltonics, Bremen, Germany) followed by mixing matrix solution consisted of α-cyano-4-hydroxycinnamic acid (4 mg/ml) in 70% acetonitrile with 0.1% TFA. The chips were then dried at room temperature and subjected into a Bruker AutoFlex MALDI-TOF MS (Bruker Daltonics, Bremen, Germany). The mass spectrometer was operated under 19 kV accelerating voltage in the reflectron mode and the m/z range from 600–4,000. The monoisotopic peptide masses obtained from MALDI-TOF MS were analyzed by m/z software. Mass spectra were internally calibrated with peptides arising from trypsin autoproteolysis at m/z = 842.509 and 2,211.105 to reach a typical mass measurement accuracy of 100 ppm.
Database searches
The peptide mass fingerprinting (PMF) data obtained from MALDI-TOF-MS was used to search for protein candidates using MASCOT software 1.0 (Matrix Science, London, UK) against the NCBInr Homo sapiens protein database. Program used to create the peak list was software flexanalysis v.2.0 and PeakErazor v.2.01. Tryptic autolytic fragments and cytokeratins were removed from the set of data by using PeakErazor software v.2.01. Justification of score threshold was 64. Moreover, identification of positive protein was based on the following criteria: (1) basis of at least six matching peptides were subjected for mass analysis; (2) at least 15% of the peptide coverage of the theoretical sequences matched.
Differential proteins verified by Western blotting
Antibodies
Antibodies against following proteins were used as first antibody: human α Enolase poly-Ab, human S100A10 poly-Ab, human SOD2 poly-Ab, human peroxiredoxin 2 poly-Ab (Santa Cruz), human S100A4 poly-Ab, human Cathepsin D poly-Ab, and human Annexin I poly-Ab (NeoMarkers).
Secondary antibodies, either anti-goat immunoglobulin G (IgG) or anti-rabbit IgG conjugated with horseradish peroxidase, were purchased from Santa Cruz, CA, USA.
Western blotting analysis
The total cell lysate was prepared in cold lysis buffer (RIPA, Santa Cruz, CA, USA) with freshly added protease inhibitor (PMSF, Santa Cruz, CA, USA), and the lysate was collected by centrifugation at 14,000×g for 15 min at 4°C. For Western blot analysis, 20 μg protein/well was electrophoresised over 15% (for S100A4) and 12% (for others) Tris-glycine polyacrylamide gels (Bio-Rad) and then transferred onto the polyvinylidene fluoride membranes. The membranes were incubated with appropriate primary antibody in blocking buffer at 4°C for overnight. After a brief washing, the membranes were incubated with appropriate secondary antibody conjugated with horseradish peroxidase (Santa Cruz) and detected by enhanced chemiluminescence (Amersham Biosciences).
Statistical analysis
Results are presented as mean ± SE. Data were analyzed by single factor analysis of variance (SPSS10.0). Differences were considered statistically significant if p < 0.05.
Results
2-DE protein patterns
The cultured SFs were morphologically fibroblast-like. 2-DE gels of synovial fibroblast protein resemble each other closely (Figs. 1 and 2). Approximately 800–900 spots on each gel could be visualized after silver staining.

Comparison of 2-DE protein patterns between healthy SFs (a) and RA SFs (b) (pH 3–10, NL, 12% T). Approximately 900 spots were visualized. Proteins of interest were identified by MALDI-TOF-MS. Two proteins were successfully identified. They are indicated by names at their spot positions in the gels. These proteins showed much higher expression levels in RA than in healthy control

The 2-DE gels (pH 4–7, 12%T) of SFs obtained from healthy control (a), RA (b), and OA(c). Master gel of the proteome of SFs in three groups was present as d. Approximately 800 spots are visualized. Twenty-five proteins derived from gels of SFs in RA and 15 proteins from those of OA among differential spots were identified by MALDI-TOF-MS. These proteins were significantly higher in SFs from RA and OA than in SFs from healthy individuals based on PDQuest V.7.3.1 analysis. Twenty-three proteins are indicated by names at their spot positions in the master gel
A set of two-group (healthy and RA groups, pH 3–10) or three-group (healthy, RA, and OA groups, pH 4–7) gels were analyzed. Spot detection and matching of these gels were done using the automated detection and matching dialog in PDQuest v 7.3.1 (Bio-Rad). After manual editing, a match set was created in which the protein spots from different gels are matched to each other and are included in a synthetic image, called a match set standard (master gel). This artificial composite image contains all the information the software needed to analyze the gels quantitatively, qualitatively, or statistically. The comparison either between healthy and RA, between healthy and OA, or between OA and RA was proceeded. Identified spots were indicated by their corresponding protein names.
Identification of differentially expressed proteins
By PMF strategy, we identified 34 proteins derived from gels of SFs in RA and 15 proteins from those of OA among differential spots. The quantity of these proteins was significantly higher in SFs from RA and OA than that from healthy individuals based on PDQuest v.7.3.1 analysis. Twenty-five proteins were obtained.
The intensity of each protein spot for RA, OA, and control samples is shown in Figs. 1 and 2. Figure 2d showed the “master gel” generated by the PDQuest software, which contains valid spots. All the proteins identified in this work were listed in Table 1. These proteins included enzymatic and structural proteins (ENOA, TPIS, LDHB, Annexin, Galectin, Lamin A/C, CRABP2, SNAPA), signal transduction proteins (GDIR, TCTP), calcium binding protein (S100A4, S100A10), etc. Most of these proteins were related to either RA or OA inflammation.
Some differentially proteins expression in SFs
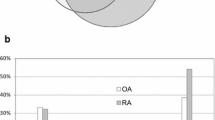
Western blot was used to further verify differential proteins in order to ensure the reliability of the proteome results from 2-DE. The results showed that expression of Enolase α, S100A4, S100A10, Annexin I (Anx-1), Cathepsin D, mitochondrial superoxide dismutase (MnSOD), peroxiredoxin 2 (PRX2) was significantly higher in SFs from both RA and OA than that from healthy individuals. Evidently, these results were consistent with proteome analysis. Furthermore, it was found that MnSOD and Cathepsin D were significantly upregulated in OA than in RA (Fig. 3).

Test of identified proteins from healthy, RA, and OA SFs by Western blot; 20 μg total protein was loaded onto the gel. After blotting, the membrane was probed with either anti-enolase α, anti-s100 A4, anti-s100 A10, anti-annexin I, anti-cathepsin D, anti-MnSOD, or anti-PRX2 antibody. Densitometry of the blots is shown at the left. The mean intensity of corresponding protein expression in various groups is shown in the right figures. The significance of the differences between the two groups is indicated by *(p < 0.05) and # (p < 0.05)
Discussion
A total of 49 spots were significantly overexpressed in either RA or OA in comparison to healthy control and were subjected to MALDI-TOF-MS. Among the spots, 25 proteins were successfully identified. The function of these proteins was involved in cell organization, energy, protein fate, cell stress, and metabolism, as described in Kumar’s work [6]. While, some unreported proteins were found in our study, i.e., S100A4, S100A10, proteasome activator PA28 beta chain, Ribosomal protein P0, Ribonucleoprotein La, and GDP dissociation inhibitors. Most importantly, our data proved that these total 25 proteins were significantly and quantitatively higher in SFs from RA and OA than that from healthy individuals. The most abundant differential proteins in SFs from RA and OA were cathepsin D, α-enolase, MnSOD, PRX2, Anx-1, and S100 calcium-binding protein A4 and A10.
Over the last 25 years, many autoantibody activities in RA have been described. Some of the RA-specific antibodies might be very helpful in the early diagnosis and prognosis of the disease [9]. The protein α-Enolase, a highly conserved multifunctional protein, is known to be upregulated by hypoxia and by proinflammatory stimuli, both of which are features of the synovial membrane microenvironment in RA. Antibodies against α-enolase have been found in the sera of RA, and citrullinated α-enolase has been described as a candidate autoantigen in RA [10]. Recently, triosephosphate isomerase (TPI) was demonstrated to be a pathogenic autoantigen in OA [11], and the anti-TPI autoantibodies were detected in a subset of patients with RA [12]. Our proteomics study show that α-enolase and TPI were upregulated in synovial fibroblasts from RA and OA, which is consistent with the results obtained from other routine methods. How an antibody against ubiquitous cytoplasmic enzyme to provoke joint-specific autoimmune disease should be further explained. But, our results indicated that α-enolase and TPI likely have potential diagnostic and prognostic value for RA.
Glucocorticoids (GCs) have been used as anti-inflammatory drugs for more than half a century. Anx-1, 37-kd member of the annexin superfamily of calcium and phospholipid binding proteins, has been suggested to function as a cellular mediator of the anti-inflammatory actions of GCs [13]. Moreover, recent evidences show that Anx-1 exerts endogenous anti-inflammatory effects on adjuvant-induced arthritis via the regulation of cytokine gene expression in the synovium. Endogenous Anx-1 is able to downregulate proinflammatory cytokines, such as IL-1β, TNFα, IL-6, and MIF expression [14]. Thus, it hints that Anx-1 could be one of the promising new agents for treatments specifically tailored to RA, and administration of Anx-1 analogs, or non-GC manipulation of endogenous Anx-1 expression, may be a valuable strategy against arthritis and other inflammatory diseases.
Increasing evidence demonstrated an important role of oxidative stress in the development of the chronic and destructive inflammation, such as RA or OA [15]. The overproduction of reactive oxygen species (ROS) molecules may contribute to the degradation of cartilage matrix [16]. To prevent toxicity by ROS, mammalian cells possess a well-coordinated antioxidant enzyme system. Examples of antioxidant enzymes are the superoxide dismutases (SOD), glutathione peroxidase, catalase, and a novel family of peroxidases, the peroxiredoxins (PRX). Increased SOD activity and increased expression of PRX5 has been reported in OA [17]. The present study show that MnSOD and PRX2 are more abundant in SFs of RA or OA, which suggests that they may play a protective role against oxidative stress in inflammatory arthritis. Furthermore, antioxidants and oxidative enzymes have been shown to ameliorate arthritis in animal models [18]. MnSOD that is upregulated to a higher level in OA than that in RA implied that inadequate control of ROS had been involved in the pathophysiology of RA. An understanding of the complex interactions involved in these pathways may allow the development of novel therapeutic strategies for RA.
It is now well understood that SFs contribute significantly to matrix degradation in RA through the expression of matrix degrading enzymes such as matrix metalloproteinases and cathepsins [19]. Cathepsins can be divided into three subgroups according to their active-site amino acid (i.e., cysteine B, C, H, F, K, L O, S, V, and W), aspartic (D and E), or serine (G) cathepsins. The cysteine proteases cathepsin B and cathepsin L, particularly, have been shown to be activated by proto-oncogenes and upregulated in RA synovium at sites of cartilage invasion [20]. The aspartate protease cathepsin D has not been studied in much detail with respect to arthritis in contrast to cathepsins B and L. However, cathepsin D has been purified from RA synovium and has been demonstrated in RA and OA synovial fluid [21]. Cathepsin D has also been found to be overexpressed in OA’s SFs as compared with that in RA’s SFs in our experiment. Thus, it is reasonable to think that cathepsin D plays an important role in the pathological alteration of joints. The correlation between cathepsin D expression and disease severity and the contribution of cathepsin D to fibroblast-mediated collagen degradation remains to be fully elucidated.
The question of whether the massive synovial hyperplasia characteristic for RA is mainly due to the proliferation of SFs or to defective apoptosis of SFs is still a matter for discussion. Over recent years, evidence has been accumulated for both hypotheses [22]. On the basis of the well-known phenomenon that SFs can grow very fast over multiple passages in vitro, cell proliferation has been considered the major mechanism for the hyperplastic growth of the RA synovium. S100A4 and S100A10 are members of the S100 calcium-binding protein family. To date, it has been demonstrated in many studies that S100A4 is involved in cancer growth and metastasis [23]. An increased expression of S100A4 mRNA has been confirmed in the lining and sublining layer of RA synovium [24]. Since several phenomena are similar between RA and malignant tumors [25], these data implied that S100A4 might be contribute to the aggressive, invasive, and tumor-like behavior of RA synovium. The mechanisms that associate S100A4 with synovitis are not well understood. The dysregulation of the transcription factors such as activator protein (AP-1), NF-κB, and the tumor suppressor gene, p53, have been implicated to play the role in the aggressive proliferative and invasive functions of SFs. The accumulation of p53 has been observed in SFs from rheumatoid patients, and an important role of p53 in regulation of cellular proliferation in rheumatoid synovium was supported by some experiments [26, 27]. This implicated that S100A4 might trigger a cascade of events through interaction with the intracellular target proteins such as tumor suppressor p53 to participate in the major mechanism for the hyperplastic growth of SFs.
In conclusion, the overexpressed proteins we found in SFs from RA and OA were included in enzymatic and structural proteins, signal transduction proteins, calcium binding protein, etc. With literature searches, our results indicated that (1) S100A4 might interact with the intracellular target proteins, such as tumor suppressor p53, to participate in the hyperplastic growth of SFs, (2) cathepsin D might play an important role in the pathological alteration of joints, (3) α-Enolase and TPI likely have potential diagnostic and prognostic value for RA, and (4) antioxidants and oxidative enzymes, as well as enhancing endogenous Anx-1 expression, might be the valuable strategies against arthritis. This proteomics analysis may significantly promote our understanding of the molecular biology of arthritis and will facilitate further studies on searching for markers of diagnosis and new therapeutic agents.
References
Hwang SY, Kim JY, Kim KW et al (2004) IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther 6:R120–R128
Malemud CJ (2007) Growth hormone, VEGF and FGF: Involvement in rheumatoid arthritis. Clinica Chimica Acta 375:10–19
Tran CN, Lundy SK, Fox DA (2005) Synovial biology and T cells in rheumatoid arthritis. Pathophysiology 12:183–189
Muller-Ladner U, Kriegsmann J, Franklin BN et al (1996) Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol 149:1607–1615
Dieppe PA, Lohmander LS (2005) Pathogenesis and management of pain in osteoarthritis. Lancet 365:965–973
Dasuri K, Antonovici M, Chen K et al (2004) The synovial proteome: analysis of fibroblast-like synoviocytes. Arthritis Res Ther 6(2):R161–168
Arnett FC, Edworthy SM, Bloch DA et al (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31:315–324
Altman R, Asch E, Bloch D et al (1986) Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum 29:1039–1049
Nell VP, Machold KP, Stamm TA et al (2005) Autoantibody profiling as early diagnostic and prognostic tool for rheumatoid arthritis. Ann Rheum Dis 64:1731–1736
Kinloch A, Tatzer V, Wait R et al (2005) Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther 7:R1421–R1429
Xiang Y, Sekine T, Nakamura H et al (2004) Proteomic surveillance of autoimmunity in osteoarthritis: identification of triosephosphate isomerase as an autoantigen in patients with osteoarthritis. Arthritis Rheum 50:1511–1521
Schaller M, Burton DR, Ditzel HJ (2001) Autoantibodies to GPI in rheumatoid arthritis: linkage between an animal model and human disease. Nat Immunol 2:746–753
Clark AR (2007) Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol 275:79–97
Yang YH, Morand EF, Getting SJ et al (2004) Modulation of inflammation and response to dexamethasone by Annexin 1 in antigen-induced arthritis. Arthritis Rheum 50:976–984
Hitchon CA, El-Gabalawy HS (2004) Oxidation in rheumatoid arthritis. Arthritis Res Ther 6:265–278
Regan E, Flannelly J, Bowler R et al (2005) Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis Rheum 52:3479–3491
Wang MX, Wei A, Yuan J et al (2002) Expression and regulation of peroxiredoxin 5 in human osteoarthritis. FEBS Lett 531:359–362
Tastekin N, Aydogdu N, Dokmeci D et al (2007) Protective effects of l-carnitine and alpha-lipoic acid in rats with adjuvant arthritis. Pharmacol Res 56:303–310
Rannou F, Francois M, Corvol MT et al (2006) Cartilage breakdown in rheumatoid arthritis. Joint Bone Spine 73:29–36
Baici A, Lang A, Zwicky R et al (2004) Cathepsin B in osteoarthritis: uncontrolled proteolysis in the wrong place. Seminars in Arthritis and Rheumatism 34:24–28
Taubert H, Riemann D, Kehlen A et al (2002) Expression of cathepsin B, D and L protein in juvenile idiopathic arthritis. Autoimmunity 35:221–224
Palao G, Santiago B, Galindo MA et al (2006) Fas activation of a proinflammatory program in rheumatoid synoviocytes and its regulation by FLIP and caspase 8 signaling. Arthritis Rheum 54:1473–1481
Hemandas AK, Salto-Tellez M, Maricar SH et al (2006) Metastasis-associated protein S100A4–a potential prognostic marker for colorectal cancer. J Surg Oncol 93:498–503
Masuda K, Masuda R, Neidhart M et al (2002) Molecular profile of synovial fibroblasts in rheumatoid arthritis depends on the stage of proliferation. Arthritis Res 4:R8
Senolt L, Grigorian M, Lukanidin E et al (2006) S100A4 (Mts1): is there any relation to the pathogenesis of rheumatoid arthritis? Autoimmun Rev 5:129–131
Salvador G, Sanmarti R, Garcia-Peiro A et al (2005) p53 expression in rheumatoid and psoriatic arthritis synovial tissue and association with joint damage. Ann Rheum Dis 64:183–187
Simelyte E, Rosengren S, Boyle DL et al (2005) Regulation of arthritis by p53: critical role of adaptive immunity. Arthritis Rheum 52:1876–1884
Acknowledgements
This work was supported by the funding of Chongqing Key Laboratory for Diseases Proteomics and partly by NSFC (30672174, 30500447).
Disclosures
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
G.-P. Bo, L.-N. Zhou and W.-F. He contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bo, GP., Zhou, LN., He, WF. et al. Analyses of differential proteome of human synovial fibroblasts obtained from arthritis. Clin Rheumatol 28, 191–199 (2009). https://doi.org/10.1007/s10067-008-1013-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-008-1013-y