
Abstract
Oxygen metabolism has an important role in the pathogenesis of rheumatoid arthritis. Reactive oxygen species (ROS) produced in the course of cellular oxidative phosphorylation, and by activated phagocytic cells during oxidative bursts, exceed the physiological buffering capacity and result in oxidative stress. The excessive production of ROS can damage protein, lipids, nucleic acids, and matrix components. They also serve as important intracellular signaling molecules that amplify the synovial inflammatory–proliferative response. Repetitive cycles of hypoxia and reoxygenation associated with changes in synovial perfusion are postulated to activate hypoxia-inducible factor-1α and nuclear factor-κB, two key transcription factors that are regulated by changes in cellular oxygenation and cytokine stimulation, and that in turn orchestrate the expression of a spectrum of genes critical to the persistence of synovitis. An understanding of the complex interactions involved in these pathways might allow the development of novel therapeutic strategies for rheumatoid arthritis.
Similar content being viewed by others


Introduction
Molecular oxygen is essential for the survival of all aerobic organisms. Aerobic energy generation is dependent on oxidative phosphorylation, a process by which the oxidoreduction energy of mitochondrial electron transport is converted to the high-energy phosphate bond of ATP. In this multi-step enzymatic process, oxygen serves as the final electron acceptor for cytochrome c oxidase, the terminal component of the mitochondrial enzymatic complex that catalyzes the four-electron reduction of O2 to H2O. A byproduct of this process is the production of partly reduced oxygen metabolites that are highly reactive and that leak out of the mitochondria and react rapidly with other molecules. In turn, reactive nitrogen species, sulfur-centered radicals, and other reactive species are generated by interactions with these molecules. Reactive oxygen species (ROS) participate in several physiological functions, and form an integral part of the organism's defense against invading microbial agents.
Because of their potentially damaging effects, several antioxidant mechanisms have evolved to protect cells and organisms from damage by excessive amounts of these highly reactive mediators. Oxidative stress is a term that is used to describe situations in which the organism's production of oxidants exceeds the capacity to neutralize them. The result can be damage to cell membranes, lipids, nucleic acids, proteins, and constituents of the extracellular matrix such as proteoglycans and collagens.
Extended periods of hypoxia, or brief periods of complete anoxia, invariably lead to death. In contrast, cellular hypoxia occurs frequently, both physiologically and pathologically, and serves as a potent stimulus for changes in gene transcription, translation, and several post-translational protein modifications that serve to rapidly adapt cells and tissues to this stimulus. Oxygen levels vary considerably in different tissues – and even in different areas of a single tissue – and depend on a complex interaction of physiological variables, particularly the balance between the vascular supply and the metabolic demands of the tissue. Hypoxia serves as a particularly potent stimulus for angiogenesis in most tissues.
In this review we explore the role of oxidative stress and hypoxia in the pathogenesis of rheumatoid arthritis (RA), a prototypical chronic inflammatory disorder, focusing on recent developments in this area, and highlighting mechanisms that can potentially be exploited therapeutically. An understanding of these processes in the context of RA has been greatly aided by knowledge gained in the areas of cancer and cardiovascular biology.
ROS in health and disease
Generation of ROS
Phagocytic cells such as macrophages and neutrophils, on activation, undergo an oxidative burst that produces highly toxic ROS that are designed to kill the invading pathogens (reviewed in [1, 2]). This oxidative burst is mediated by the NADPH oxidase system, and results in a marked increase in oxygen consumption and the production of superoxide (O2-•). NADPH is composed of several subunits that assemble at the plasma membrane and fuse with intracellular phagocytic vesicles or the outer membrane. This allows the concentrated release of oxidants formed subsequently. Superoxide is converted to hydrogen peroxide (H2O2) either spontaneously or more rapidly when catalyzed by superoxide dismutatase, an enzyme that occurs in two isoforms, one of which is inducible by inflammatory cytokines such as tumor necrosis factor-α (TNF-α).
In the presence of ferrous ions (Fe2+) and other transition metals, hydrogen peroxide and superoxide are converted via the Fenton reaction to highly reactive, aqueous soluble hydroxyl radicals (OH•) that are probably responsible for much of the cell toxicity associated with ROS. Additionally, the neutrophil-associated enzyme myeloperoxidase can oxidize halides such as chloride (Cl-) and convert hydrogen peroxide into hypochlorous acid (HOCl), which then can interact with amino acids to form chloramines. Similar reactions can occur with other halides such as bromide and iodide. Further reaction of hydrogen peroxide with hypochlorous acid produces singlet oxygen, another highly reactive and damaging radical. Reactions of hypochlorous acid with amino acids lead to aldehyde production. Superoxide can also react with nitric oxide (NO), synthesized from the deimination of L-arginine by nitric oxide synthase (NOS), and produce the highly reactive peroxynitrite radical (ONOO-). These reactions are summarized in Table 1.
Physiological roles for ROS
ROS are produced during normal aerobic cell metabolism, have important physiological roles in maintaining cell redox status, and are required for normal cellular metabolism including intracellular signaling pathways and the activity of transcription factors such as NF-κB, activator protein 1 and hypoxia-inducible factor-1α (HIF-1α) (see below). In addition, ROS produced by phagocytes also seem to have important physiological roles in priming the immune system. A functional mutation of a component of the NADPH oxidase complex, Ncf1, produces a lower oxidative burst and enhanced arthritis susceptibility and severity in murine pristane-induced arthritis [3, 4]. Activation of the NADPH complex by vitamin E ameliorated arthritis when given before arthritis induction, indicating that the Ncf1 functional polymorphism is involved at the immune priming stage of disease. The authors of those papers propose that the physiological production of ROS by phagocytes in response to antigen affects T cell–antigen interactions and possibly induces apoptosis of autoreactive arthritogenic T cells, thereby preventing autoimmune responses. In humans, Ncf1 is redundant and a complete loss of function is associated with chronic granulomatous disease that has increased susceptibility to microbial infections. The associations of Ncf1 with other experimental autoimmune conditions suggest that polymorphisms in the Ncf1 gene might be important for autoimmunity in general [5].
Oxidant defense mechanisms
Several defense mechanisms have evolved to protect cellular systems from oxidative damage. These include intracellular enzymes such as superoxide dismutase, glutathione peroxidase, catalase and other peroxidases, thioredoxin reductase, the sequestration of metal ion cofactors such as Fe and Cu by binding to proteins, and endogenous antioxidants. Superoxide dismutase (SOD) enhances the otherwise slow spontaneous breakdown of superoxide, forming the less toxic hydrogen peroxide, which can then interact with glutathione and ultimately form H2O and O2. SOD exists in a constitutively expressed form and an inducible form (MnSOD) that resides in mitochondria. MnSOD is induced by cytokines through NF-κB and may require other cofactors including nucleolar phosphosmin, an RNA-binding protein [6]. Glutathione peroxidase, the primary mitochondrial defense from hydrogen peroxide, is upregulated by p53 and hypoxia [7, 8]. Catalase also degrades hydrogen peroxide, and probably has a function in cytosolic or extracellular protection from oxidants because it is absent from the mitochondria of most cells. The thioredoxin–thioredoxin reductase system is another essential component of the cellular response to oxidative stress, especially in cardiac tissue [9]. Several stressors, including inflammatory cytokines and oxidative stress, induce thioredoxin. Thioredoxin regulates protein redox status and, when activated, facilitates protein–DNA interactions. In cardiac tissue, thioredoxin expression is enhanced under conditions of cyclic hypoxia and reperfusion. Enhanced thioredoxin expression has also been demonstrated in RA synovial fluid and tissue [10–12].
Endogenous antioxidants protect cellular systems from the damaging effects of ROS and reactive nitrogen species (RNS) reviewed in [13]. The main antioxidants are vitamin A (retinol and metabolites), vitamin C (ascorbic acid) and vitamin E (α-tocopherol). β-Carotene, a water-soluble provitamin A, is a free-radical scavenger that controls the propagation of reactive species and influences lipoxygenase activity. Vitamin C (ascorbic acid), one of the first lines of defense from oxidative stress, can prevent lipid peroxidation by trapping water-soluble peroxyl radicals before their diffusion into lipid membranes; it also reacts with superoxide, peroxy, and hydroxyl radicals, and is important in recycling other antioxidants such as vitamin E. Vitamin E has lipid-soluble properties that allow it to act as a chain-breaking reagent in lipid peroxidation.
Evidence for oxidative stress in RA
Several lines of evidence suggest a role for oxidative stress in the pathogenesis of RA. Epidemiologic studies have shown an inverse association between dietary intake of antioxidants and RA incidence [14–17], and inverse associations between antioxidant levels and inflammation have been found [18, 19]. Iron, a catalyst for hydroxyl radical production from hydrogen peroxide (see Table 1), is present in RA synovial tissue and is associated with poorer prognosis [20]. Several groups have demonstrated increased oxidative enzyme activity along with decreased antioxidant levels in RA sera and synovial fluids [21–25]. Because of the highly reactive nature of ROS, it is difficult to directly demonstrate their presence in vivo. It is considerably more practical to measure the 'footprints' of ROS and RNS, such as their effects on various lipids, proteins, and nucleic acids. Thus, evidence for oxidative stress in RA has in many cases been generated by approaches that detect oxidant-induced changes to these molecules (reviewed in [1, 26–28]). Studies of RA synovial fluid and tissue have demonstrated oxidative damage to hyaluronic acid [29], lipid peroxidation products [30, 31], oxidized low-density-lipid proteins (LDL) [32], and increased carbonyl groups reflective of oxidation damage to proteins [32, 33]. Evidence of oxidative damage to cartilage, extracellular collagen, and intracellular DNA has also been demonstrated (see below). Oxidative stress has been shown to induce T cell hyporesponsiveness in RA through effects on proteins and proteosomal degradation [34]. Finally, antioxidants and oxidative enzymes have been shown to ameliorate arthritis in animal models [35–37].
Cartilage/collagen effects
ROS and RNS damage cellular elements in cartilage directly and damage components of the extracellular matrix either directly or indirectly by upregulating mediators of matrix degradation (reviewed in [2, 26]). Modification of amino acids by oxidation, nitrosylation, nitration, and chlorination can alter protein structure and impair biological function, leading to cell death. ROS impair chondrocyte responses to growth factors and migration to sites of cartilage injury; RNS, in particular NO, interfere with interactions between chondrocytes and the extracellular matrix [38]. NO can also increase chondrocyte apoptosis.
Oxygen and nitrogen radicals inhibit the synthesis of matrix components including proteoglycans by chondrocytes. In particular, NO and O2 seem to inhibit type II collagen and proteoglycan synthesis and the sulfation of newly synthesized glycosaminoglycans. Oxygen radicals can cause low levels of collagen fragmentation and enhanced collagen fibril cross-linking. Oxygen radicals have also been shown to fragment hyaluronan and chondroitin sulfate [39, 40] and damage the hyaluronan-binding region of the proteoglycan core protein, thereby interfering with proteoglycan–hyaluronan interactions [41]. In addition, ROS and RNS can damage the components of the extracellular matrix indirectly through the activation and upregulation of matrix metalloproteinases.
Oxidative damage to immunoglobulins – advanced glycation end-products
Oxidative stress occurring during inflammation can cause proteins to become non-enzymatically damaged by glyoxidation. This process, which involves primarily lysine and arginine residues, ultimately results in the generation of advanced glycation endproducts (AGE), which are stable. An example of this process is the glyoxidation of hemoglobin to hemoglobin A1c in the context of repetitive hyperglycemia. The immunoglobin molecule can also undergo similar glyoxidation to generate AGE-IgG. In the context of inflammatory arthritis, we have shown that antibodies to AGE-IgG are specifically associated with RA, whereas the actual formation of AGE-IgG is related to the intensity of the systemic inflammatory response, and is not specific to RA [42, 43].
Genotoxic effects of oxidative stress
Reactive oxygen and nitrogen species directly damage DNA and impair DNA repair mechanisms. This damage can occur in the form of DNA strand breakage or individual nucleotide base damage. DNA reaction products, in particular 8-oxo-7-hydro-deoxyguanosine formed by the reaction of hydroxyl radicals (OH) with deoxyguanosine, are elevated in leukocytes and sera of patients with RA [44, 45]. This product is particularly mutagenic and cytotoxic. NO, especially in high concentrations, causes the deamination of deoxynucleotides, DNA strand breakage and oxidative damage from peroxynitrite, and DNA modification by metabolically activated N-nitrosamines, all of which can lead to somatic mutations.
RA tissue has evidence of microsatellite instability reflecting ongoing mutagenesis [46]. Such mutagenesis is normally corrected by DNA repair systems including the mismatch repair (MMR) system; however, the MMR system is defective in RA, probably due in part to oxidative stress. Evidence for this comes from findings of decreased expression of hMSH6, a component of the MutSα complex that is important for repair of the single base mismatches that are characteristic of oxidative stress, and increased expression of hMSH3, a component of MutSβ that is important for the repair of insertion or deletion loops. This pattern of MMR expression was reproduced by synovial fibroblasts exposed to reactive nitrate species and to a smaller extent by fibroblasts exposed to ROS, indicating a role for oxidative stress in the development of microsatellite instability in RA. The authors of this work suggest that this pattern of MMR expression might allow short-term cell survival by preventing potentially major DNA damage at the expense of minor DNA damage or that it might promote the development of a mutated phenotype having additional survival benefit.
Although somatic DNA mutations probably occur randomly through the genome, they may occur in the coding regions of functional genes. An example of this is the p53 tumor suppressor gene. The p53 tumor suppressor protein is important in containing and repairing mutations through its effects on growth regulating genes, G1 growth arrest, interactions with DNA repair mechanisms, and apoptosis. In addition, wild-type p53 downregulates NOS and subsequent NO production through interaction with the region of the NOS2 promoter [47]. Somatic mutations of p53 have been demonstrated in RA synovium and cultured RA fibroblast-like synoviocytes [48, 49], and have been implicated in the pathogenesis of inflammatory arthritis [28]. These are primarily transitional mutations consistent with mutations resulting from oxidative deamination by nitric oxide or oxygen radicals, and are similar to those found in tumors. Importantly, there is a distinct geographical distribution of the mutations in RA synovium [50]. The distribution of p53 mutations was patchy, with most being located in the lining layer, an area distant from oxygenating vasculature and bathed in oxidant-rich synovial fluid. Specific histologic correlation was not provided; however, it is interesting to speculate that the areas with a high frequency of p53 mutations might also have lining layer hyperplasia and that these mutations contribute to the formation of the invasive pannus.
Mitochondrial DNA (mtDNA) is particularly susceptible to oxidative stress, and prolonged exposure leads to persistent mtDNA damage without effective repair, loss of mitochondrial function, cell growth arrest, and apoptosis [51]. This increased susceptibility probably relates to the proximity of mtDNA to oxidative reactive species including the lipid peroxidation products generated from inner mitochondrial membrane lipids, which contain components of the respiratory electron transport chain, or a lack of protecting histones, or potentially inefficient repair mechanisms. The relevance of mtDNA to inflammatory arthritis is found from studies demonstrating that extracellular mtDNA is increased in RA synovial fluid and plasma [45] and that oxidatively damaged mtDNA can induce murine arthritis [52].
Lipid peroxidation
Lipid peroxidation has been implicated in the pathogenesis of cancer, atherosclerosis, degenerative diseases, and inflammatory arthritis. During lipid peroxidation, polyunsaturated fatty acids are oxidized to produce lipid peroxyl radicals that in turn lead to further oxidation of polyunsaturated fatty acid in a perpetuating chain reaction that can lead to cell membrane damage (see Table 1). Matrix degradation arising from cytokine-stimulated chondrocytes was shown to be primarily due to lipid peroxidation, and to be preventable by vitamin E, the primary antioxidant for lipids [53].
Lipid oxidation probably contributes to accelerated atherosclerosis in RA [54–56]. Persistent local and systemic elevation of inflammatory cytokines promotes lipolysis, and the systemic release of free fatty acids contributes to the dyslipidemia seen in RA. Oxidative stress arising from inflammatory reactions leads to the oxidation of local LDL. Oxidized LDL promotes further inflammatory changes, including local upregulation of adhesion molecules and chemokines. Advanced glycation endproducts might also contribute to this inflammation. Monocytes ingest large quantities of oxidized LDL, resulting in the formation of foam cells that are present in atherosclerotic plaques of vessels and have also been found in RA synovial fluid [57] and synovium [58].
Role of hypoxia and reoxygenation in RA synovitis
Several lines of evidence have suggested that cycles of hypoxia/reoxygenation are important in sustaining RA synovitis. It has long been known that RA synovial fluids are hypoxic, acidotic, and exhibit low glucose and elevated lactate concentrations [59, 60]. This biochemical profile is indicative of anaerobic metabolism in the synovium [61, 62]. We have recently repeated the seminal experiments evaluating pO2 levels in RA synovial fluids and found that the pO2 levels are frequently below those detected in venous blood, with some being as low as 10 mmHg (CAH and HSE-G, unpublished work). These levels correlated with lactic acid levels. It has proven more difficult to measure pO2 levels in RA synovium directly in vivo. Two studies, published in abstract form, evaluated RA synovial pO2 with microelectrodes and found these levels to be quite low [63, 64]. These data are supported by similar findings in experimental inflammatory arthritis [65]; together they support the notion that RA synovitis has the features of a chronically hypoxic microenvironment that compensates by using anaerobic metabolism.
Cellular responses to hypoxia: the role of HIF-1α
The potential role of hypoxia in RA synovitis has largely been extrapolated from studies of tumors, in which the rapidly proliferative state and high metabolic demands of the tumor cells result in areas of hypoxia generated by an imbalance between the demands and the abnormal tumor vascular supply. This hypoxic microenvironment potently stimulates tumor angiogenesis and results in phenotypic changes in the tumor cells that favor survival and growth in this environment [66, 67]. The biological basis of this process has been well studied, and relates to the exquisite regulation of a key transcription factor, HIF-1α [68]. This oxygen-sensitive transcription factor orchestrates the expression of a wide spectrum of genes that serve, first, to allow the cells to use anaerobic metabolism to generate energy; second, to enhance survival and inhibit apoptosis; and third, to improve the supply of oxygen by promoting angiogenesis and increased oxygen-carrying capacity.
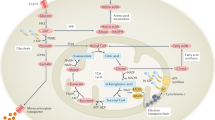
In view of the crucial role of HIF-1α in cellular adaptation to hypoxia, its regulation needs to be rapidly responsive to changes in the cellular oxygen supply. Although several mechanisms have been proposed for oxygen sensing, it has been shown that the primary mechanism by which hypoxia directly regulates HIF-1α is by inhibiting its degradation [68]. Under aerobic conditions HIF-1α is undetectable because of a rapid process of ubiquitination and subsequent proteosomal degradation. This degradative process is mediated by von Hippel–Landau tumor suppressor factor (VHL) [69, 70], which when mutated results in von Hippel–Landau syndrome, characterized by the formation of hemangiomas due to uninhibited angiogenesis. The interaction between HIF-1α and VHL requires the critical hydroxylation of two proline residues (402 and 564) and one asparagine residue (803), as well as the acetylation of a lysine residue (532) in HIF-1α [71, 72]. The hydroxylation events are mediated by a family of three prolyl hydroxylases (PHD-1, PHD-2, and PHD-3) and one asparagine hydroxylase (FIH), and require O2 and several cofactors, particularly iron and ascorbate (Fig. 1). In the absence of O2, this critical hydroxylation becomes rate limiting, thus preventing HIF-1α from being degraded and leaving it free to bind to its constitutively expressed partner, HIF-1β (aryl hydrocarbon nuclear translocator; ARNT).

Hypoxic regulation of the hypoxia-inducible factor-1α (HIF-1α) transcription factor is primarily through inhibition of degradation. Under normoxic conditions, HIF-1α undergoes rapid proteosomal degradation once it forms a complex with von Hippel–Landau tumor suppressor factor (VHL) and E3 ligase complex. This requires the hydroxylation of critical proline residues by a family of HIF-1α-specific prolyl hydroxylases (PHD-1,2,3), which requires O2 and several cofactors, including iron. Under hypoxic conditions, or when iron is chelated or competitively inhibited, proline hydroxylation does not occur, thus stabilizing HIF-1α and allowing it to interact with the constitutively expressed HIF-1β (aryl hydrocarbon nuclear translocator; ARNT). The HIF-1 complex then translocates to the nucleus and activates genes with hypoxia-responsive elements in their promoters. bHLH, basic helix-loop-helix; CBP, cAMP response element binding protein; FIH, factor inhibiting HIF-1α; PAS, PER-ARNT-SIM; TAD, transactivation domain.
It should be noted that the degradation of HIF-1α can also be inhibited by approaches that limit the availability of iron. Thus, cobalt chloride (CoCl2), a competitive inhibitor, and desferioxamine, an iron chelator, both potently stabilize HIF-1α in vitro and mimic the effects of hypoxia. HIF-1α/ARNT form a complex with CBP/p300, and this complex rapidly translocates to the nucleus and transactivates genes that have a hypoxia-responsive element (HRE) in their promoters featuring the consensus motif RCGTG. Although the full complement of HRE-regulated genes are obviously present in all cells, the hypoxia-induced expression of some of these genes, such as erythropoietin, is quite tissue specific. Other genes, such as vascular endothelial growth factor (VEGF), and genes encoding for glycolytic enzymes, are induced by hypoxic stimulation in most cells. It is interesting to speculate that glucose-6-phosphate isomerase, which as been proposed as an autoantigen in RA [73–75], is induced by hypoxia in a HIF-1α-dependent manner [76]. The list of genes that have been shown to be directly regulated by HIF-1α is shown in Fig. 2.

Genes that have been shown to be directly regulated by hypoxia-inducible factor-1α through hypoxia-responsive elements in their promoter regions. The genes are classified on the basis on their best known functional properties. A full listing of the gene annotations is presented in the Additional file.
Thus, although there is now a well-defined group of genes that are regulated by hypoxia through HIF-1α, their patterns of expression vary in different cells and tissues. Interestingly, it has recently been demonstrated that HIF-1α is essential for the function of myeloid cells of the innate immune systems such as neutrophils and macrophages [77]. This study demonstrated that the regulation of glycolytic capacity by HIF-1α in these myeloid cells is crucial for the energy generation required for cell aggregation, motility, invasiveness, and bacterial killing. Of particular relevance to RA was the marked attenuation of synovitis and articular damage in an adjuvant arthritis model when HIF-1α was absent.
The effects of ROS on HIF-1α itself have been controversial [78]. One hypothesis suggests that ROS are produced by the NADPH oxidase system and serve to inhibit HIF-1α activation [79]. During hypoxia, reduced ROS formation serves to activate HIF-1α by diminished inhibition. An alternative hypothesis suggests that ROS are in fact produced by mitochondria during hypoxia and may indeed serve to stabilize HIF-1α and promote nuclear localization and gene transcription [80, 81]. There is experimental evidence in support of both of these competing hypotheses, and indeed, both may be correct depending on the intensity and duration of the hypoxic stimulus, and on the cell type involved.
In addition to hypoxic regulation of HIF-1α, it has been established that cytokines and growth factors such as interleukin-1β (IL-1β), TNF-α, transforming growth factor-β (TGF-β), platelet-derived growth factor, fibroblast growth factor-2, and insulin-like growth factors are capable of stabilizing and activating this key transcription factor under normoxic conditions [82–87]. Several signaling pathways are involved, particularly the phosphoinositide 3-kinase (PI-3K)/Akt pathway, and the mitogen-activated protein (MAP) kinase pathway. It is likely that the normoxic regulation of HIF-1α by the PI-3K/Akt pathway involves increased translation of the protein, whereas MAP kinase regulation involves phosphorylation of the molecule, which in turn increases its transactivating capacity [88, 89]. The regulation of HIF-1α by NO has also recently been shown to be mediated by the MAP kinase and PI-3K/Akt pathways [89].
HIF-1α and hypoxia-regulated genes in RA synovitis
The expression of HIF-1α has been evaluated in RA and other forms of synovitis [90–92]. One study suggested that HIF-1α is widely expressed in RA synovium, and on the basis of evaluating consecutive sections it was assumed to be expressed in a cytoplasmic pattern by macrophages in both the lining and sublining areas [92]. A second study evaluated the expression of HIF-1α and the related protein HIF-2α in RA, osteoarthritis, and normal synovium, and found them to be widely expressed in both RA and osteoarthritis but not in normal synovium [90]. The synovial expression of HIF-1α in this study was in a mixed nuclear and cytoplasmic pattern, and was seen in most lining cells, stromal cells, mononuclear cells, and blood vessels. On the basis of these findings, the authors suggested a role for hypoxia and HIF-1α in the pathogenesis of both RA and osteoarthritis.
Our own studies of synovial HIF-1α expression have suggested a more limited, patchy pattern of nuclear expression that was confined primarily to the lining cells of RA tissues with a particularly hyperplastic lining layer [91] (Fig. 3). Indeed, when we exposed fresh synovial tissue explants to hypoxic culture conditions, the nuclear expression of HIF-1α increased markedly in the lining cell layer, in a manner analogous to that seen in cultured synovial fibroblasts. It should be noted that our immunohistology studies were performed on snap-frozen sections of synovium with the use of three commercially available anti-HIF-1α antibodies. In contrast, the two other studies used archival synovial tissue that had been deparaffinized and then subjected to antigen retrieval techniques. It is currently not clear whether these technical considerations are sufficient to explain these discrepant findings.

Expression of hypoxia-inducible factor-1α (HIF-1α) in RA synovium and fibroblast-like synoviocytes under normoxic and hypoxic conditions. (a) Under normoxic conditions, HIF-1α expression in fresh synovial explants was patchy and confined to some cells in the lining layer. (b) When fresh RA tissue explants were cultured in hypoxic conditions (1% O2), nuclear staining for HIF-1α was readily detected in the lining cells. (c, d) A similar pattern of expression was seen in fibroblast-like synoviocytes where under normoxic conditions no HIF-1α staining was detected (c), whereas under hypoxic conditions intense nuclear staining was seen maximally at 4–6 hours (d). Reproduced, with permission, from [91].
The presence of regional HIF-1α expression in hyperplastic areas of the RA lining layer would be consistent with a dynamic process in which the lining cells in these areas, being the furthest removed from a precarious and insufficient vascular supply in the sublining areas, are subjected to fluctuating oxygen levels, resulting in repetitive cycles of hypoxia and reperfusion. Moreover, such a regional distribution of HIF-1α expression would also be in keeping with the known rapid stabilization and nuclear translocation of HIF-1α under transient hypoxic conditions, which is followed by equally rapid degradation of this transcription factor when relative normoxia is re-established [93].
The expression of several HIF-1α-regulated genes has been explored in RA synovitis, in particular angiogenesis mediators such as VEGF and the angiopoietins. VEGF has been shown to be upregulated in the serum, synovial fluid, and synovium of patients with RA [94–98]. Moreover, clinical response to TNF-α inhibitors is associated with a decrease in systemic and synovial VEGF levels, this being attributed to inhibition of synovial angiogenesis [96, 99]. At the cellular level, the regulation of VEGF expression is complex. We and others have shown that cytokines abundant in RA synovium, such as TNF-α, IL-1β, and TGF-β, interact with hypoxia in an additive manner to induce VEGF expression by fibroblast-like synoviocytes [91, 100]. The interaction at the level of the VEGF promoter between HIF-1α and SMAD3, the latter being the mediator of TGF-β transcriptional regulation, has been demonstrated [101]. Similarly, the angiopoietins Ang1 and Ang2, and their cellular receptor Tie2, which are all widely expressed in RA synovitis, are regulated by both hypoxia and TNF-α [102–105]. These observations underscore the complexity of transcriptional regulation in a chronic inflammatory microenvironment such as RA synovium, and indicate that the regulation of specific genes by hypoxia occurs in the context of multiple other regulatory pathways, particularly the NF-κB pathway.
Hypoxia, or hypoxia and reoxygenation?
Studies of RA synovium in vivo have suggested that synovial perfusion is influenced directly by high intraarticular pressures that are further increased by movement [106–108]. On the basis of these observations, it can therefore be proposed that intermittent joint loading with ambulation, especially in the setting of an effused joint, enhances local joint hypoxia, which in turn is followed by reoxygenation when the joint is unloaded. A predicted consequence of such cycles of hypoxia and reoxygenation would be cycles of HIF-1α expression and the genes it regulates, followed by repetitive bursts of ROS formation. The ROS generated serve as a stimulus for NF-κB activation, probably through effects on upstream kinases [109, 110]. This includes effects on the dissociation of NF-κB from its inhibitor IκB (which requires oxidation), the regulation of IκB degradation, and the binding of NF-κB to DNA (which requires a reducing environment). Activation of NF-κB serves to induce the expression of multiple proinflammatory genes, many of which are also regulated by HIF-1α [78, 111]. This interaction is summarized in Fig. 4. The resultant changes in gene and protein expression are complex and vary in different cell types, but overall can be expected to promote inflammation, angiogenesis, and enhanced cell survival, all cardinal features of RA synovitis.

Regulation of the hypoxia-inducible factor-1α (HIF-1α) and nuclear factor-κB (NF-κB) pathways by reactive oxygen species (ROS) and cytokine stimulation. The complex and interrelated activation of these two critical transcription factors is central to most of the processes that sustain synovitis in rheumatoid arthritis, such as endothelial activation, leukocyte recruitment, angiogenesis, and enhanced cell survival. IL, interleukin; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; TNF, tumor necrosis factor.
The sequelae of hypoxia and reoxygenation have been addressed in vascular models, and some limited experimental evidence has addressed this question in RA synovium [112]. Interestingly, the vascular models of hypoxia and reoxygenation have demonstrated a phenomenon that has been termed preconditioning. This describes a process whereby a cell or a tissue becomes resistant to subsequent hypoxic episodes after transient exposure to a hypoxic episode. The biological basis of preconditioning continues to be defined, and might involve signaling by Akt [113] and/or extracellular signal-related kinase 1/2 [114], and possibly an upregulation of PHD-2 during the hypoxic phase [115]. It is currently not known whether some form of preconditioning occurs in RA synovitis, and whether this promotes the survival of cells in this oxidatively stressed microenvironment.
Therapeutic considerations
Targeting ROS with antioxidants
Various forms of antioxidant therapy have demonstrated promising results in experimental arthritis models [35–37]. The polyphenolic fraction of green tea containing potent antioxidants prevents collagen-induced arthritis [116]. The beneficial effects seem to be due to the catechin epigallocatechin-3-gallate (EGCG), which inhibits IL-1β-mediated inflammatory effects, including NOS and NO production by human chondrocytes [117], and inhibits MMP activity [118, 119].
There is widespread availability and interest in the use of antioxidant supplementation by patients with inflammatory arthritis, although proof of efficacy is modest. A traditional Mediterranean diet relatively high in antioxidants improved RA disease activity and functional status after 3 months compared with a standard 'Western' diet, although clinical improvement was not associated with any significant change in plasma levels of antioxidants [16, 120]. In a separate study of patients with RA, supplementation with antioxidants vitamin A, E, and C increased plasma antioxidant levels with a corresponding decrease in malondialdehyde, a marker of oxidative stress; however, a clinical response was not reported [121]. Specific supplementation of oral vitamin E, the major lipid-soluble antioxidant in human plasma, erythrocytes, and tissue, had no effect on RA disease activity or indices of inflammation but did improve pain, suggesting a role in central analgesia mechanisms [122].
Targeting angiogenesis
It has been proposed that the formation of destructive RA pannus is dependent on synovial angiogenesis, in a manner analogous to locally invasive tumors. As is the case with many tumors, hypoxia has a central role in regulating this angiogenic process. On this basis, inhibition of synovial angiogenesis has been proposed as a rational therapeutic strategy, and several angiogenesis inhibitors have been shown to have favorable effects in animal models (reviewed in [123]). As mentioned earlier, it has been suggested that the therapeutic responses to TNF-α inhibition might be attributable, at least in part, to an inhibition of angiogenesis [99].
An alternative hypothesis suggests that, rather than representing a tumor-like proliferative process that outgrows its vascular supply, RA pannus represents a non-healing synovial wound that is prevented from resolution by an inadequate vascular supply. Hypoxia has long been proposed as an important stimulus in wound healing [124]. Moreover, hypoxia and HIF-1α serve to stimulate genes that are involved in wound repair and the formation of granulation tissue, a process critically dependent on angiogenesis [125–129]. Interestingly, the expression of HIF-1α protein does not occur during the initial inflammatory process but becomes evident within 1–5 days of wounding, and seems to have a prominent role in the subsequent tissue healing. If RA synovitis does have many of the features of a non-healing wound, inhibition of angiogenesis would conceptually not represent an appropriate strategy and indeed might have deleterious effects, depending on the stage of the synovitis being treated.
Targeting HIF-1α and hypoxic cells
Our understanding of cellular and tissue responses to changes in oxygen tension has increased markedly over the past decade. The central role of HIF-1α in mediating hypoxic responses has suggested new therapeutic opportunities, particularly in cancer and cardiovascular medicine [130, 131]. Small molecules targeting the HIF-1α pathway are currently being developed and show considerable promise in cancer models. It should be noted that many cancer cells overexpress HIF-1α on a genetic basis, a phenomenon that presumably enhances their survival in hypoxic environments [131]. It is not clear whether an analogous situation exists in RA pannus. As mentioned above, studies evaluating the expression of HIF-1α in RA synovitis have not provided a consistent picture, although all studies so far have pointed to the synovial lining layer as the main site of HIF-1α expression. It is not clear whether this expression is 'physiological', in response to poor tissue oxygenation, or pathological, as seen in many tumors. Moreover, chondrocytes that function in a physiologically hypoxic environment are critically dependent on HIF-1α for normal development and maintenance of cartilage integrity [132–136]. Thus, targeting HIF-1α in an articular disorder such as RA remains a conceptually challenging proposition requiring considerably more experimental data.
An alternative approach is to target hypoxic cells by using their 'reducing' intracellular microenvironment to generate toxic metabolites locally from specific drugs [137]. These 'bioreductive' drugs would thus be more toxic to hypoxic than normoxic cells. Alternatively, such drugs could serve as carriers for delivering anti-inflammatory compounds to target tissues. One such bioreductive drug, metronidazole, has been proposed as potentially being useful for this purpose, although a controlled clinical trial had produced mostly disappointing results [138].
Conclusions
Repetitive cycles of hypoxia and reoxygenation, along with oxidants produced by phagocytic cells such as macrophages and neutrophils, lead to chronic oxidative stress in the RA synovial microenvironment. The ROS that are generated damage proteins, nucleic acids, lipids, and matrix components, and serve to amplify signaling pathways that sustain the synovitis. HIF-1α and NF-κB are key transcription factors that respond to changes in cellular oxygenation and that orchestrate the expression of a spectrum of genes that are critical to the persistence of the synovitis. An understanding of the complex interactions involved in these pathways may allow the development of novel therapeutic strategies for RA.
Abbreviations
- AGE:
-
advanced glycation endproducts
- HIF-1α:
-
hypoxia-inducible factor-1α
- LDL:
-
low-density-lipid proteins
- MAP:
-
mitogen-activated protein
- MMR:
-
mismatch repair
- mtDNA:
-
mitochondrial DNA
- NF-κB:
-
nuclear factor-κB
- PHD:
-
prolyl hydroxylase
- PI-3K:
-
phosphoinositide 3-kinase
- RA:
-
rheumatoid arthritis
- RNS:
-
reactive nitrogen species
- ROS:
-
reactive oxygen species
- SOD:
-
superoxide dismutase
- TGF:
-
transforming growth factor
- TNF:
-
tumor necrosis factor
- VEGF:
-
vascular endothelial growth factor
- VHL:
-
von Hippel–Landau tumor suppressor factor.
References
Babior BM: Phagocytes and oxidative stress. Am J Med. 2000, 109: 33-44. 10.1016/S0002-9343(00)00481-2.
Lotz M: Neuropeptides, free radicals and nitric oxide. Rheumatology. 2003, 135-146.
Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, Holmdahl R: Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet. 2003, 33: 25-32. 10.1038/ng1058.
van de Loo FA, Bennink MB, Arntz OJ, Smeets RL, Lubberts E, Joosten LA, van Lent PL, Coenen-de Roo CJ, Cuzzocrea S, Segal BH, et al: Deficiency of NADPH oxidase components p47phox and gp91phox caused granulomatous synovitis and increased connective tissue destruction in experimental arthritis models. Am J Pathol. 2003, 163: 1525-1537.
van der Veen RC, Dietlin TA, Hofman FM, Pen L, Segal BH, Holland SM: Superoxide prevents nitric oxide-mediated suppression of helper T lymphocytes: decreased autoimmune encephalomyelitis in nicotinamide adenine dinucleotide phosphate oxidase knockout mice. J Immunol. 2000, 164: 5177-5183.
Dhar SK, Lynn BC, Daosukho C, St Clair DK: Identification of nucleophosmin as an NF-kB co-activator for the induction of the human SOD2 gene. J Biol Chem. 2004, 279: 28209-28219. 10.1074/jbc.M403553200.
Tan M, Li S, Swaroop M, Guan K, Oberley LW, Sun Y: Transcriptional activation of the human glutathione peroxidase promoter by p53. J Biol Chem. 1999, 274: 12061-12066. 10.1074/jbc.274.17.12061.
Bierl C, Voetsch B, Jin RC, Handy DE, Loscalzo J: Determinants of human plasma glutathione peroxidase (GPx-3) expression. J Biol Chem. 2004, 279: 26839-26845. 10.1074/jbc.M401907200.
Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, Wagner T, Vatner SF, Sadoshima J: Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. 2003, 112: 1395-1406. 10.1172/JCI200317700.
Turoczi T, Chang VW, Engelman RM, Maulik N, Ho YS, Das DK: Thioredoxin redox signaling in the ischemic heart: an insight with transgenic mice overexpressing Trx1. J Mol Cell Cardiol. 2003, 35: 695-704. 10.1016/S0022-2828(03)00117-2.
Das DK: Thioredoxin regulation of ischemic preconditioning. Antioxid Redox Signal. 2004, 6: 405-412. 10.1089/152308604322899477.
Maurice MM, Nakamura H, Gringhuis S, Okamoto T, Yoshida S, Kullmann F, Lechner S, van der Voort EA, Leow A, Versendaal J, Muller-Ladner U, Yodoi J, Tak PP, Breedveld FC, Verweij CL: Expression of the thioredoxin-thioredoxin reductase system in the inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42: 2430-2439. 10.1002/1529-0131(199911)42:11<2430::AID-ANR22>3.0.CO;2-6.
Sowers M, Lachance L: Vitamins and arthritis. The roles of vitamins A, C, D, and E. Rheum Dis Clin North Am. 1999, 25: 315-332.
Cerhan JR, Saag KG, Merlino LA, Mikuls TR, Criswell LA: Antioxidant micronutrients and risk of rheumatoid arthritis in a cohort of older women. Am J Epidemiol. 2003, 157: 345-354. 10.1093/aje/kwf205.
Heliovaara M, Knekt P, Aho K, Aaran RK, Alfthan G, Aromaa A: Serum antioxidants and risk of rheumatoid arthritis. Ann Rheum Dis. 1994, 53: 51-53.
Hagfors L, Leanderson P, Skoldstam L, Andersson J, Johansson G: Antioxidant intake, plasma antioxidants and oxidative stress in a randomized, controlled, parallel, Mediterranean dietary intervention study on patients with rheumatoid arthritis. Nutr J. 2003, 2: 5-10.1186/1475-2891-2-5.
Bae SC, Kim SJ, Sung MK: Inadequate antioxidant nutrient intake and altered plasma antioxidant status of rheumatoid arthritis patients. J Am Coll Nutr. 2003, 22: 311-315.
Paredes S, Girona J, Hurt-Camejo E, Vallve JC, Olive S, Heras M, Benito P, Masana L: Antioxidant vitamins and lipid peroxidation in patients with rheumatoid arthritis: association with inflammatory markers. J Rheumatol. 2002, 29: 2271-2277.
Mulherin DM, Thurnham DI, Situnayake RD: Glutathione reductase activity, riboflavin status, and disease activity in rheumatoid arthritis. Ann Rheum Dis. 1996, 55: 837-840.
Dabbagh AJ, Trenam CW, Morris CJ, Blake DR: Iron in joint inflammation. Ann Rheum Dis. 1993, 52: 67-73.
De Leo ME, Tranghese A, Passantino M, Mordente A, Lizzio MM, Galeotti T, Zoli A: Manganese superoxide dismutase, glutathione peroxidase, and total radical trapping antioxidant capacity in active rheumatoid arthritis. J Rheumatol. 2002, 29: 2245-2246.
Taysi S, Polat F, Gul M, Sari RA, Bakan E: Lipid peroxidation, some extracellular antioxidants, and antioxidant enzymes in serum of patients with rheumatoid arthritis. Rheumatol Int. 2002, 21: 200-204. 10.1007/s00296-001-0163-x.
Cimen MY, Cimen OB, Kacmaz M, Ozturk HS, Yorgancioglu R, Durak I: Oxidant/antioxidant status of the erythrocytes from patients with rheumatoid arthritis. Clin Rheumatol. 2000, 19: 275-277.
Marklund SL, Bjelle A, Elmqvist LG: Superoxide dismutase isoenzymes of the synovial fluid in rheumatoid arthritis and in reactive arthritides. Ann Rheum Dis. 1986, 45: 847-851.
Ozturk HS, Cimen MY, Cimen OB, Kacmaz M, Durak I: Oxidant/antioxidant status of plasma samples from patients with rheumatoid arthritis. Rheumatol Int. 1999, 19: 35-37. 10.1007/s002960050097.
Henrotin YE, Bruckner P, Pujol JP: The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003, 11: 747-755. 10.1016/S1063-4584(03)00150-X.
Halliwell B: Oxygen radicals, nitric oxide and human inflammatory joint disease. Ann Rheum Dis. 1995, 54: 505-510.
Tak PP, Zvaifler NJ, Green DR, Firestein GS: Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000, 21: 78-82. 10.1016/S0167-5699(99)01552-2.
Grootveld M, Henderson EB, Farrell A, Blake DR, Parkes HG, Haycock P: Oxidative damage to hyaluronate and glucose in synovial fluid during exercise of the inflamed rheumatoid joint. Detection of abnormal low-molecular-mass metabolites by proton-n.m.r. spectroscopy. Biochem J. 1991, 273: 459-467.
Rowley D, Gutteridge JM, Blake D, Farr M, Halliwell B: Lipid peroxidation in rheumatoid arthritis: thiobarbituric acid-reactive material and catalytic iron salts in synovial fluid from rheumatoid patients. Clin Sci (Lond). 1984, 66: 691-695.
Taysi S, Polat F, Gul M, Sari RA, Bakan E: Lipid peroxidation, some extracellular antioxidants, and antioxidant enzymes in serum of patients with rheumatoid arthritis. Rheumatol Int. 2002, 21: 200-204. 10.1007/s00296-001-0163-x.
Dai L, Lamb DJ, Leake DS, Kus ML, Jones HW, Morris CJ, Winyard PG: Evidence for oxidised low density lipoprotein in synovial fluid from rheumatoid arthritis patients. Free Radic Res. 2000, 32: 479-486.
Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R: Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003, 329: 23-38. 10.1016/S0009-8981(03)00003-2.
Cemerski S, van Meerwijk JP, Romagnoli P: Oxidative-stress-induced T lymphocyte hyporesponsiveness is caused by structural modification rather than proteasomal degradation of crucial TCR signaling molecules. Eur J Immunol. 2003, 33: 2178-2185. 10.1002/eji.200323898.
Bandt MD, Grossin M, Driss F, Pincemail J, Babin-Chevaye C, Pasquier C: Vitamin E uncouples joint destruction and clinical inflammation in a transgenic mouse model of rheumatoid arthritis. Arthritis Rheum. 2002, 46: 522-532. 10.1002/art.10085.
Cuzzocrea S, McDonald MC, Mota-Filipe H, Mazzon E, Costantino G, Britti D, Mazzullo G, Caputi AP, Thiemermann C: Beneficial effects of tempol, a membrane-permeable radical scavenger, in a rodent model of collagen-induced arthritis. Arthritis Rheum. 2000, 43: 320-328. 10.1002/1529-0131(200002)43:2<320::AID-ANR11>3.0.CO;2-9.
Venkatraman JT, Chu WC: Effects of dietary omega-3 and omega-6 lipids and vitamin E on serum cytokines, lipid mediators and anti-DNA antibodies in a mouse model for rheumatoid arthritis. J Am Coll Nutr. 1999, 18: 602-613.
Clancy RM, Rediske J, Tang X, Nijher N, Frenkel S, Philips M, Abramson SB: Outside-in signaling in the chondrocyte. Nitric oxide disrupts fibronectin-induced assembly of a subplasmalemmal actin/rho A/focal adhesion kinase signaling complex. J Clin Invest. 1997, 100: 1789-1796.
Rees MD, Hawkins CL, Davies MJ: Hypochlorite-mediated fragmentation of hyaluronan, chondroitin sulfates, and related N-acetyl glycosamines: evidence for chloramide intermediates, free radical transfer reactions, and site-specific fragmentation. J Am Chem Soc. 2003, 125: 13719-13733. 10.1021/ja0370591.
Rees MD, Hawkins CL, Davies MJ: Hypochlorite and superoxide radicals can act synergistically to induce fragmentation of hyaluronan and chondroitin sulfates. Biochem J. 2004, 381: 175-184. 10.1042/BJ20040148.
Panasyuk A, Frati E, Ribault D, Mitrovic D: Effect of reactive oxygen species on the biosynthesis and structure of newly synthesized proteoglycans. Free Radic Biol Med. 1994, 16: 157-167. 10.1016/0891-5849(94)90139-2.
Newkirk MM, LePage K, Niwa T, Rubin L: Advanced glycation endproducts (AGE) on IgG, a target for circulating antibodies in North American Indians with rheumatoid arthritis (RA). Cell Mol Biol (Noisy-le-grand). 1998, 44: 1129-1138.
Newkirk MM, Goldbach-Mansky R, Lee J, Hoxworth J, McCoy A, Yarboro C, Klippel J, El Gabalawy HS: Advanced glycation end-product (AGE)-damaged IgG and IgM autoantibodies to IgG-AGE in patients with early synovitis. Arthritis Res Ther. 2003, 5: R82-R90. 10.1186/ar622.
Bashir S, Harris G, Denman MA, Blake DR, Winyard PG: Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann Rheum Dis. 1993, 52: 659-666.
Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV: Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther. 2003, 5: R234-R240. 10.1186/ar787.
Lee SH, Chang DK, Goel A, Boland CR, Bugbee W, Boyle DL, Firestein GS: Microsatellite instability and suppressed DNA repair enzyme expression in rheumatoid arthritis. J Immunol. 2003, 170: 4869-
Forrester K, Ambs S, Lupold SE, Kapust RB, Spillare EA, Weinberg WC, Felley-Bosco E, Wang XW, Geller DA, Tzeng E, et al: Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc Natl Acad Sci USA. 1996, 93: 2442-2447. 10.1073/pnas.93.6.2442.
Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR: Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 1997, 94: 10895-10900. 10.1073/pnas.94.20.10895.
Inazuka M, Tahira T, Horiuchi T, Harashima S, Sawabe T, Kondo M, Miyahara H, Hayashi K: Analysis of p53 tumour suppressor gene somatic mutations in rheumatoid arthritis synovium. Rheumatology (Oxford). 2000, 39: 262-266.
Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS: Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 2002, 99: 10025-10030. 10.1073/pnas.152333199.
Yakes FM, Van Houten B: Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997, 94: 514-519. 10.1073/pnas.94.2.514.
Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A: Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004, 75: 995-1000. 10.1189/jlb.0703328.
Tiku ML, Shah R, Allison GT: Evidence linking chondrocyte lipid peroxidation to cartilage matrix protein degradation. Possible role in cartilage aging and the pathogenesis of osteoarthritis. J Biol Chem. 2000, 275: 20069-20076. 10.1074/jbc.M907604199.
Lusis AJ: Atherosclerosis. Nature. 2000, 407: 233-241. 10.1038/35025203.
del Rincon I, Escalante A: Atherosclerotic cardiovascular disease in rheumatoid arthritis. Curr Rheumatol Rep. 2003, 5: 278-286.
Sattar N, McCarey DW, Capell H, McInnes IB: Explaining how 'high-grade' systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation. 2003, 108: 2957-2963. 10.1161/01.CIR.0000099844.31524.05.
Dai L, Lamb DJ, Leake DS, Kus ML, Jones HW, Morris CJ, Winyard PG: Evidence for oxidised low density lipoprotein in synovial fluid from rheumatoid arthritis patients. Free Radic Res. 2000, 32: 479-486.
Winyard PG, Tatzber F, Esterbauer H, Kus ML, Blake DR, Morris CJ: Presence of foam cells containing oxidised low density lipoprotein in the synovial membrane from patients with rheumatoid arthritis. Ann Rheum Dis. 1993, 52: 677-680.
Treuhaft PS, McCarty DJ: Synovial fluid pH, lactate, oxygen and carbon dioxide partial pressure in various joint diseases. Arthritis Rheum. 1971, 14: 475-484.
Lund-Olesen K: Oxygen tension in synovial fluids. Arthritis Rheum. 1970, 13: 769-776.
Naughton D, Whelan M, Smith EC, Williams R, Blake DR, Grootveld M: An investigation of the abnormal metabolic status of synovial fluid from patients with rheumatoid arthritis by high field proton nuclear magnetic resonance spectroscopy. FEBS Lett. 1993, 317: 135-138. 10.1016/0014-5793(93)81508-W.
Naughton DP, Haywood R, Blake DR, Edmonds S, Hawkes GE, Grootveld M: A comparative evaluation of the metabolic profiles of normal and inflammatory knee-joint synovial fluids by high resolution proton NMR spectroscopy. FEBS Lett. 1993, 332: 221-225. 10.1016/0014-5793(93)80636-9.
Taylor P, Miotla JM, Etherington P, Winlove P, Young Y, Paleolog E, Maini RN: VEGF release is associated with hypoxia in inflammatory arthritis [abstract]. Arthritis Rheum. 2000, 43 (Suppl 9): S296-
Ellis GA, Edmonds SE, Gaffney K, Williams RB: Synovial tissue oxygenation profile in inflamed and non-inflamed knee joints [abstract]. Br J Rheumatol. 1994, 33 (Suppl 1): 172-
Miotla J, Maciewicz R, Kendrew J, Feldmann M, Paleolog E: Treatment with soluble VEGF receptor reduces disease severity in murine collagen-induced arthritis. Lab Invest. 2000, 80: 1195-1205.
Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, et al: Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998, 394: 485-490. 10.1038/28867.
Marx J: Cell biology. How cells endure low oxygen. Science. 2004, 303: 1454-1456. 10.1126/science.303.5663.1454.
Semenza GL: HIF-1, O2, and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001, 107: 1-3. 10.1016/S0092-8674(01)00518-9.
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG: Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat Cell Biol. 2000, 2: 423-427. 10.1038/35017054.
Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH: Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000, 275: 25733-25741. 10.1074/jbc.M002740200.
Wenger RH: Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16: 1151-1162. 10.1096/fj.01-0944rev.
Distler JH, Wenger RH, Gassmann M, Kurowska M, Hirth A, Gay S, Distler O: Physiologic responses to hypoxia and implications for hypoxia-inducible factors in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2004, 50: 10-23. 10.1002/art.11425.
Matsumoto I, Staub A, Benoist C, Mathis D: Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999, 286: 1732-1735. 10.1126/science.286.5445.1732.
Schaller M, Burton DR, Ditzel HJ: Autoantibodies to GPI in rheumatoid arthritis: linkage between an animal model and human disease. Nat Immunol. 2001, 2: 746-753. 10.1038/90696.
Matsumoto I, Lee DM, Goldbach-Mansky R, Sumida T, Hitchon CA, Schur PH, Anderson RJ, Coblyn JS, Weinblatt ME, Brenner M, et al: Low prevalence of antibodies to glucose-6-phosphate isomerase in patients with rheumatoid arthritis and a spectrum of other chronic autoimmune disorders. Arthritis Rheum. 2003, 48: 944-954. 10.1002/art.10898.
Naughton DP: Hypoxia-induced upregulation of the glycolytic enzyme glucose-6-phosphate isomerase perpetuates rheumatoid arthritis. Med Hypotheses. 2003, 60: 332-334. 10.1016/S0306-9877(02)00396-1.
Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al: HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003, 112: 645-657. 10.1016/S0092-8674(03)00154-5.
Michiels C, Minet E, Mottet D, Raes M: Regulation of gene expression by oxygen: NF-κB and HIF-1, two extremes. Free Radic Biol Med. 2002, 33: 1231-1242. 10.1016/S0891-5849(02)01045-6.
Huang LE, Willmore WG, Gu J, Goldberg MA, Bunn HF: Inhibition of hypoxia-inducible factor 1 activation by carbon monoxide and nitric oxide. Implications for oxygen sensing and signaling. J Biol Chem. 1999, 274: 9038-9044. 10.1074/jbc.274.13.9038.
Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT: Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000, 275: 25130-25138. 10.1074/jbc.M001914200.
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT: Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998, 95: 11715-11720. 10.1073/pnas.95.20.11715.
Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL: Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002, 277: 38205-38211. 10.1074/jbc.M203781200.
Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L: IL-1β-mediated up-regulation of HIF-1α via an NFκB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17: 2115-2117.
Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Burgel T: Normoxic induction of the hypoxia-inducible factor 1α by insulin and interleukin-1β involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512: 157-162. 10.1016/S0014-5793(02)02247-0.
Zhou J, Schmid T, Brune B: Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB-dependent pathway. Mol Biol Cell. 2003, 14: 2216-2225. 10.1091/mbc.E02-09-0598.
Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L: Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor κB activation. Biochem J. 2003, 370: 1011-1017. 10.1042/BJ20021279.
Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E: Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem J. 2000, 350: 307-312. 10.1042/0264-6021:3500307.
Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, Remacle J, Michiels C: ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000, 468: 53-58. 10.1016/S0014-5793(00)01181-9.
Kasuno K, Takabuchi S, Fukuda K, Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL, Hirota K: Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem. 2004, 279: 2550-2558. 10.1074/jbc.M308197200.
Giatromanolaki A, Sivridis E, Maltezos E, Athanassou N, Papazoglou D, Gatter KC, Harris AL, Koukourakis MI: Upregulated hypoxia inducible factor-1α and -2α pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2003, 5: R193-R201. 10.1186/ar756.
Hitchon CA, Wong K, Ma G, Reed J, Lyttle D, El Gabalawy H: Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002, 46: 2587-2597. 10.1002/art.10520.
Hollander AP, Corke KP, Freemont AJ, Lewis CE: Expression of hypoxia-inducible factor 1α by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001, 44: 1540-1544. 10.1002/1529-0131(200107)44:7<1540::AID-ART277>3.0.CO;2-7.
Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassmann M: Induction of HIF-1α in response to hypoxia is instantaneous. FASEB J. 2001, 15: 1312-1314.
Wauke K, Nagashima M, Ishiwata T, Asano G, Yoshino S: Expression and localization of vascular endothelial growth factor-C in rheumatoid arthritis synovial tissue. J Rheumatol. 2002, 29: 34-38.
Kasama T, Shiozawa F, Kobayashi K, Yajima N, Hanyuda M, Takeuchi HT, Mori Y, Negishi M, Ide H, Adachi M: Vascular endothelial growth factor expression by activated synovial leukocytes in rheumatoid arthritis: critical involvement of the interaction with synovial fibroblasts. Arthritis Rheum. 2001, 44: 2512-2524. 10.1002/1529-0131(200111)44:11<2512::AID-ART431>3.0.CO;2-O.
Ballara S, Taylor PC, Reusch P, Marme D, Feldmann M, Maini RN, Paleolog EM: Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis Rheum. 2001, 44: 2055-2064. 10.1002/1529-0131(200109)44:9<2055::AID-ART355>3.0.CO;2-2.
Pufe T, Petersen W, Tillmann B, Mentlein R: Splice variants VEGF121 and VEGF165 of the angiogenic peptide vascular endothelial cell growth factor are expressed in the synovial tissue of patients with rheumatoid arthritis. J Rheumatol. 2001, 28: 1482-1485.
Fava RA, Olsen NJ, Spencer-Green G, Yeo KT, Yeo TK, Berse B, Jackman RW, Senger DR, Dvorak HF, Brown LF: Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994, 180: 341-346. 10.1084/jem.180.1.341.
Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN: Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor alpha and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. 1998, 41: 1258-1265. 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1.
Berse B, Hunt JA, Diegel RJ, Morganelli P, Yeo K, Brown F, Fava RA: Hypoxia augments cytokine (transforming growth factor-β (TGF-β) and IL-1)-induced vascular endothelial growth factor secretion by human synovial fibroblasts. Clin Exp Immunol. 1999, 115: 176-182. 10.1046/j.1365-2249.1999.00775.x.
Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C: Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001, 276: 38527-38535. 10.1074/jbc.M104536200.
Takahara K, Iioka T, Furukawa K, Uchida T, Nakashima M, Tsukazaki T, Shindo H: Autocrine/paracrine role of the angiopoietin-1 and -2/Tie2 system in cell proliferation and chemotaxis of cultured fibroblastic synoviocytes in rheumatoid arthritis. Hum Pathol. 2004, 35: 150-158. 10.1016/j.humpath.2003.11.010.
Fearon U, Griosios K, Fraser A, Reece R, Emery P, Jones PF, Veale DJ: Angiopoietins, growth factors, and vascular morphology in early arthritis. J Rheumatol. 2003, 30: 260-268.
DeBusk LM, Chen Y, Nishishita T, Chen J, Thomas JW, Lin PC: Tie2 receptor tyrosine kinase, a major mediator of tumor necrosis factor alpha-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum. 2003, 48: 2461-2471. 10.1002/art.11213.
Gravallese EM, Pettit AR, Lee R, Madore R, Manning C, Tsay A, Gaspar J, Goldring MB, Goldring SR, Oettgen P: Angiopoietin-1 is expressed in the synovium of patients with rheumatoid arthritis and is induced by tumour necrosis factor alpha. Ann Rheum Dis. 2003, 62: 100-107. 10.1136/ard.62.2.100.
Stevens CR, Blake DR, Merry P, Revell PA, Levick JR: A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum. 1991, 34: 1508-1513.
Levick JR: Hypoxia and acidosis in chronic inflammatory arthritis; relation to vascular supply and dynamic effusion pressure. J Rheumatol. 1990, 17: 579-582.
James MJ, Cleland LG, Rofe AM, Leslie AL: Intraarticular pressure and the relationship between synovial perfusion and metabolic demand. J Rheumatol. 1990, 17: 521-527.
Bonizzi G, Piette J, Merville MP, Bours V: Cell type-specific role for reactive oxygen species in nuclear factor-κB activation by interleukin-1. Biochem Pharmacol. 2000, 59: 7-11. 10.1016/S0006-2952(99)00290-7.
Bonizzi G, Piette J, Schoonbroodt S, Greimers R, Havard L, Merville MP, Bours V: Reactive oxygen intermediate-dependent NF-κB activation by interleukin-1β requires 5-lipoxygenase or NADPH oxidase activity. Mol Cell Biol. 1999, 19: 1950-1960.
D'Angio CT, Finkelstein JN: Oxygen regulation of gene expression: a study in opposites. Mol Genet Metab. 2000, 71: 371-380. 10.1006/mgme.2000.3074.
Han MK, Kim JS, Park BH, Kim JR, Hwang BY, Lee HY, Song EK, Yoo WH: NF-κB-dependent lymphocyte hyperadhesiveness to synovial fibroblasts by hypoxia and reoxygenation: potential role in rheumatoid arthritis. J Leukoc Biol. 2003, 73: 525-529. 10.1189/jlb.0502256.
Uchiyama T, Engelman RM, Maulik N, Das DK: Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation. 2004, 109: 3042-3049. 10.1161/01.CIR.0000130647.29030.90.
Jones NM, Bergeron M: Hypoxia-induced ischemic tolerance in neonatal rat brain involves enhanced ERK1/2 signaling. J Neurochem. 2004, 89: 157-167.
Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E: Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-α-prolyl-4-hydroxylases. Biochem J. 2004, 381: 761-767. 10.1042/BJ20040620.
Haqqi TM, Anthony DD, Gupta S, Ahmad N, Lee MS, Kumar GK, Mukhtar H: Prevention of collagen-induced arthritis in mice by a polyphenolic fraction from green tea. Proc Natl Acad Sci USA. 1999, 96: 4524-4529. 10.1073/pnas.96.8.4524.
Singh R, Ahmed S, Islam N, Goldberg VM, Haqqi TM: Epigallocatechin-3-gallate inhibits interleukin-1β-induced expression of nitric oxide synthase and production of nitric oxide in human chondrocytes: suppression of nuclear factor κB activation by degradation of the inhibitor of nuclear factor κB. Arthritis Rheum. 2002, 46: 2079-2086. 10.1002/art.10443.
Ahmed S, Wang N, Lalonde M, Goldberg VM, Haqqi TM: Green tea polyphenol epigallocatechin-3-gallate (EGCG) differentially inhibits interleukin-1β-induced expression of matrix metalloproteinase-1 and -13 in human chondrocytes. J Pharmacol Exp Ther. 2004, 308: 767-773. 10.1124/jpet.103.059220.
Ahmed S, Rahman A, Hasnain A, Lalonde M, Goldberg VM, Haqqi TM: Green tea polyphenol epigallocatechin-3-gallate inhibits the IL-1β-induced activity and expression of cyclooxygenase-2 and nitric oxide synthase-2 in human chondrocytes. Free Radic Biol Med. 2002, 33: 1097-1105. 10.1016/S0891-5849(02)01004-3.
Skoldstam L, Hagfors L, Johansson G: An experimental study of a Mediterranean diet intervention for patients with rheumatoid arthritis. Ann Rheum Dis. 2003, 62: 208-214. 10.1136/ard.62.3.208.
Jaswal S, Mehta HC, Sood AK, Kaur J: Antioxidant status in rheumatoid arthritis and role of antioxidant therapy. Clin Chim Acta. 2003, 338: 123-129. 10.1016/j.cccn.2003.08.011.
Edmonds SE, Winyard PG, Guo R, Kidd B, Merry P, Langrish-Smith A, Hansen C, Ramm S, Blake DR: Putative analgesic activity of repeated oral doses of vitamin E in the treatment of rheumatoid arthritis. Results of a prospective placebo controlled double blind trial. Ann Rheum Dis. 1997, 56: 649-655.
Koch AE: Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003, 62 (Suppl 2): ii60-67.
Trabold O, Wagner S, Wicke C, Scheuenstuhl H, Hussain MZ, Rosen N, Seremetiev A, Becker HD, Hunt TK: Lactate and oxygen constitute a fundamental regulatory mechanism in wound healing. Wound Repair Regen. 2003, 11: 504-509. 10.1046/j.1524-475X.2003.11621.x.
Albina JE, Reichner JS: Oxygen and the regulation of gene expression in wounds. Wound Repair Regen. 2003, 11: 445-451. 10.1046/j.1524-475X.2003.11619.x.
Haroon ZA, Raleigh JA, Greenberg CS, Dewhirst MW: Early wound healing exhibits cytokine surge without evidence of hypoxia. Ann Surg. 2000, 231: 137-147. 10.1097/00000658-200001000-00020.
Ozawa K, Kondo T, Hori O, Kitao Y, Stern DM, Eisenmenger W, Ogawa S, Ohshima T: Expression of the oxygen-regulated protein ORP150 accelerates wound healing by modulating intracellular VEGF transport. J Clin Invest. 2001, 108: 41-50. 10.1172/JCI200111772.
Scheid A, Wenger RH, Christina H, Camenisch I, Ferenc A, Stauffer UG, Gassmann M, Meuli M: Hypoxia-regulated gene expression in fetal wound regeneration and adult wound repair. Pediatr Surg Int. 2000, 16: 232-236. 10.1007/s003830050735.
Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry WL Jr, Reichner JS: HIF-1 expression in healing wounds: HIF-1α induction in primary inflammatory cells by TNF-α. Am J Physiol Cell Physiol. 2001, 281: C1971-C1977.
Giaccia A, Siim BG, Johnson RS: HIF-1 as a target for drug development. Nat Rev Drug Discov. 2003, 2: 803-811. 10.1038/nrd1199.
Semenza GL: Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003, 3: 721-732. 10.1038/nrc1187.
Pufe T, Lemke A, Kurz B, Petersen W, Tillmann B, Grodzinsky AJ, Mentlein R: Mechanical overload induces VEGF in cartilage discs via hypoxia-inducible factor. Am J Pathol. 2004, 164: 185-192.
Stokes DG, Liu G, Coimbra IB, Piera-Velazquez S, Crowl RM, Jimenez SA: Assessment of the gene expression profile of differentiated and dedifferentiated human fetal chondrocytes by microarray analysis. Arthritis Rheum. 2002, 46: 404-419. 10.1002/art.10106.
Coimbra IB, Jimenez SA, Hawkins DF, Piera-Velazquez S, Stokes DG: Hypoxia inducible factor-1α expression in human normal and osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2004, 12: 336-345. 10.1016/j.joca.2003.12.005.
Rajpurohit R, Koch CJ, Tao Z, Teixeira CM, Shapiro IM: Adaptation of chondrocytes to low oxygen tension: relationship between hypoxia and cellular metabolism. J Cell Physiol. 1996, 168: 424-432. 10.1002/(SICI)1097-4652(199608)168:2<424::AID-JCP21>3.0.CO;2-1.
Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS: Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001, 15: 2865-2876.
Bodamyali T, Stevens CR, Billingham ME, Ohta S, Blake DR: Influence of hypoxia in inflammatory synovitis. Ann Rheum Dis. 1998, 57: 703-710.
Marshall DA, Hunter JA, Capell HA: Double blind, placebo controlled study of metronidazole as a disease modifying agent in the treatment of rheumatoid arthritis. Ann Rheum Dis. 1992, 51: 758-760.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Electronic supplementary material
13075_2004_1327_MOESM1_ESM.xls
Additional File 1: An Excel file containing a table that gives details of the gene annotations used in Fig. 2. (XLS 66 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Hitchon, C.A., El-Gabalawy, H.S. Oxidation in rheumatoid arthritis. Arthritis Res Ther 6, 265 (2004). https://doi.org/10.1186/ar1447
Published:
DOI: https://doi.org/10.1186/ar1447