
Abstract
This work presents the results of the investigations on the electropolishing and anodic oxidation of the Ti–13Nb–13Zr titanium alloy. Electropolishing was conducted in the solution containing ammonium fluoride and sulfuric acid, whereas the solution of phosphoric acid was used for anodic oxidation of the alloy. The influence of electropolishing and anodization process parameters on the texture (scanning electron microscopy (SEM)) and chemical composition (X-ray photoelectron spectroscopy (XPS)) of the surface layer was established. Electrochemical impedance spectroscopy in 5 % NaCl solution was used for the determination of the corrosion resistance of the alloy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Titanium exists in one of two allotropic forms, α or β, with a phase transformation temperature of 883 °C [1]. The transformation temperature can be altered by adding alloying elements. Elements such as aluminum (Al), O, and N stabilize the α phase by increasing the beta transus temperature, and the beta titanium alloys are stabilized by the isomorphous and eutectoid stabilizers such as Mo, V, Nb, Cr, Mn, Fe, Co, Ni, and Cu. Depending on the type and amount of phase stabilizers, the alloy structures are classified into three major categories: alpha (α), alpha plus beta (α + β), and beta (β) phase [2]. Titanium alloys exhibit high tensile strength, modulus of elasticity, and fatigue strength. The alloys used to produce biomaterials should show mechanical properties close to those they replace and they should not contain toxic elements; thus, the compositions of these alloys are constantly changing and evolving [3-6].
One of the most popular alloys used in medicine is Ti–6Al–4V. However, that alloy exhibits poor tribological properties, and the Young’s modulus (~110 GPa) is higher than that of the bone (~10–64 GPa) [7-9]. Metallic vanadium is thermodynamically unstable under the conditions corresponding to the tissue environment [10]. Moreover, vanadium is carcinogenic [11, 12]. Vanadium oxide (V2O5) exhibits relatively good solubility and high toxicity in the living organisms. Al has well-documented toxic effects in the serum or urine of the patients who have total hip replacements composed of the titanium alloy [13, 14]. In addition, aluminum has a causal relationship with the neurotoxicity and senile dementia of the Alzheimer type [15-17]. The increasingly stringent requirements of the long-term implants must be met, and the growing doubts regarding the cytotoxicity of the titanium alloys containing vanadium have triggered investigations on the vanadium-free titanium alloys. The Ti–13Nb–13Zr (TNZ) alloy was developed by Davidson and Kovacs [18] and is a part of the new class of biomedical alloys to be used in medical implants. It is a near-α alloy and consists of hexagonal closed-packed (hcp) martensite (α′) at the water-quenched condition. With the subsequent aging, the alloy consists of hcp martensite (α′) and the submicroscopic body-centered cubic (bcc) beta precipitates. The dispersed beta precipitates strengthen and harden the material. The aging treatment normally results in the conversion of the martensite into α + β mixture for β-Ti alloys (with bcc crystalline structure). The presence of martensite in the aged alloy indicates that the alloy is more α/β alloy than a near-β alloy. At the aged conditions, this alloy has the modulus of elasticity around 65 GPa [19].
Their high corrosion resistance titanium and its alloys owe to the presence of the oxide layer on the surface. The thickness of the oxide layer on the surface of titanium may be increased by thermal or electrochemical oxidation. Typical anodic passivation gives the oxide layers of the thickness from several tenths to several hundredths of nanometer [20]. The changes of the thickness are accompanied by the change in the color of the oxide layer. The most important parameter that influences the crystal structure and the thickness of the oxide layer is the voltage of the passivation. Below 20 V, the oxide layer is amorphous, whereas above 45 V, the oxide layer becomes crystalline, and microcrystalline forms of both anatase and rutile are visible [21, 22]. The chemical composition of the oxide layer depends on the chemical composition of the surface of the sample as well as on the electrolyte used. In the case of the Ti–6Al–4V alloy, besides TiO2, Al2O3 and V2O5 are also observed [23], whereas in the case of Ti–6Al–7Nb, the surface layer is composed of TiO2, Al2O3, and Nb2O5 [24, 25]. Despite many years of investigations on the corrosion resistance of the Ti–13Nb–13Zr alloy in different environments, new publications concerning this problem are appearing regularly [26-29]. Anodic treatment of the Ti–13Nb–13Zr alloy is not widely described in the accessible literature. Investigations on electropolishing [30], preparation of the nanotubular oxide layers [31-33], and plasma electrolytic oxidation were conducted up to now [34-38]. Preparation of thin oxide layers on the Ti–13Nb–13Zr alloy was investigated by voltammetry but up to only 8 V [39, 40].
The aim of this work was to investigate the influence of the voltage applied in the anodic oxidation on the morphology and chemical composition of the oxide layer formed on the surface of the Ti–13Nb–13Zr alloy and its corrosion resistance in the NaCl solution.
Experimental
Table 1 presents the composition of the Ti–13Nb–13Zr alloy (BIMO Metals, Wrocław, Poland) used in the investigations. The samples had cylindrical shape with the diameter of 9.5 mm and the height of 7 mm. They were ground using #320 and #600 abrasive papers (SiC) and then cleaned 5 min in an ultrasonic wash in 2-propanol and deionized water. Afterwards, the samples were polished electrochemically. The electropolishing was carried out in a 1-dm3 plastic electrolyzer, with a cover, two cathodes made of the acid-resistant steel, and an anode clamp for fixing the samples. The process was carried out with the use of the Unitra 5352M power supply. The solution temperature (25 ± 0.1 °C) was maintained with the UH-4 thermostat. The Ti–13Nb–13Zr alloy samples were polished in the bath composed of 12.5 M sulfuric acid, 5.5 M ethylene glycol, and 2.7 M ammonium fluoride [30].
After electropolishing, alloy samples were anodized in the bath containing H3PO4 (2 M). The anodization was performed during 5 min at the voltage of 20, 60, 100, or 140 V and at the maximum current density of 10 A dm−2, using a DC power supply PWR800H (Kikusui, Japan) and a cooled electrolyzer. The power supply was operated in the following manner: initially, the current was set to a constant value of 10 A dm−2 and the voltage was allowed to grow—when the assumed voltage was attained, the current was continuously automatically adjusted to the value allowing to keep the voltage constant (Fig. 1). The Ti–13Nb–13Zr alloy specimen served as the anode, and the cathode was a titanium mesh. After anodization, the samples were rinsed with distilled water and cleaned ultrasonically in 2-propanol and deionized water.

The voltage–current–time response for the Ti–13Nb–13Zr alloy sample anodized at 140 V
The morphology of sample surfaces was studied using the high-resolution scanning electron microscope Inspect F and the atomic force microscope NanoScope E (Digital Instruments). The chemical composition of the selected samples was determined using energy-dispersive X-ray spectroscopy (EDX) system attached to a scanning electron microscope (SEM) (Oxford Instruments) and by X-ray photoelectron spectroscopy (XPS) Multitechnique Electron Spectrometer PHI 5700/660 (Physical Electronics) with a cylindrical mirror analyzer. XPS spectra were measured using the monochromatic Al Kα radiation (1,486.6 eV) with the energy resolution of about 0.3 eV. Ellipsometric measurements were performed using the EP3 ellipsometer (Nanofilm). The range of wavelength was 360–1,001 nm and the angle of incidence was 80°. To calculate the thickness of the oxide film, the EM Bruggemann model was used. The cross section of the sample anodized at 140 V was examined using SEM (Hitachi S-3400N) with the EDX system (Thermo Noran).
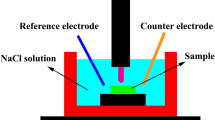
The electrochemical measurements were performed using the IM5d Impedance Spectrum Analyzer (Zahner Elektrik, Kronach, Germany). Samples were molded in an epoxy resin with wire connections attached in such a manner that only one circular flat surface was exposed to the solution. Measurements were performed in the 5 % NaCl solution in a glass cell in the three-electrode configuration. The frequency range used in the EIS measurements was 0.1 to 100,000 Hz, and the amplitude of the sinusoidal voltage signal was 5 mV. A platinum foil was used as the counter electrode, and an Ag|AgCl/3.5 M KCl electrode was used as the reference electrode. All potentials in this paper are given with respect to that electrode. During the measurement, the solution was stirred with a magnetic stirrer with the rotation speed of 15 Hz. All EIS spectra were measured at the spontaneously established corrosion potential. The first spectrum was recorded just after sample immersion in the solution then the measurement was repeated every 2 h during 1 day.
All the solutions were prepared using analytical grade reagents from POCh Gliwice, Poland, and deionized water was obtained from the Millipore-Q system.
Results and discussion
The course of the current and the voltage during the anodization of the Ti–13Nb–13Zr alloy is presented in Fig. 1. When the anodizing voltage is set to 140 V or higher, during the first stage of the process (ca. 18 s), voltage grows fast with the rate of about 7.5 V s−1, up to approximately 140 V. At that voltage, micro-arc oxidation starts, causing the breakdown of the oxide layer. After the attainment of the voltage of 140 V, the current drops down to the value below 1 mA. Compact oxide layer was obtained when the anodizing voltage was kept below 140 V. The color of the layer depends on the voltage: the layer formed at the highest voltage is pale gray, at 100 V willow green layer is formed, at 60 V the layer is yellow gold, and at the lowest voltage (20 V) the layer attains dark violet color. The electropolishing leads to the smoothing and brightening of the Ti–13Nb–13Zr alloy surface (Fig. 2) [30]. Application of the voltage lower than the breakdown voltage gives the oxide layer which replicates the morphology of the substrate [20]. Pores characteristic for the micro-arc oxidation process, formed by the breakdown of the oxide layer, appear at the surface only at the voltage of 140 V (Fig. 2) [38]. The pores are uniformly distributed over the surface and their diameter is smaller than 2 μm. The peaks of only those metals that form the substrate are visible in the EDX spectrum of the electropolished Ti–13Nb–13Zr alloy sample, whereas the spectra of the samples subjected to anodic oxidation at 100 and 140 V show also the peaks belonging to phosphorus (Fig. 2). Evidently, phosphorus is being built in the growing layer during the anodization. The relative ratio of phosphorus in the case of the sample anodized at the voltage of 140 V is higher than in the case of the sample anodized at the voltage of 100 V. The anodization at low voltage leads usually to the embedding of some components of the electrolyte in the growing layer, but their concentrations are rather low. Only at the voltages above the breakdown voltage the concentration of the built-in components becomes bigger.

The SEM and AFM images as well as EDX spectra of the Ti–13Nb–13Zr alloy samples
The chemical composition of the electropolished Ti–13Nb–13Zr alloy surface (Ti/Zr/Nb = 4.0:0.55:1) is similar to the composition of the alloy (Ti/Zr/Nb = 5.6:1:1), but the surface is depleted in titanium and zirconium. The chemical composition of the surface oxide layer formed at 100 V (Ti/Zr/Nb = 12.6:1:1) is similar to air-formed native oxide layer on that alloy (Ti/Zr/Nb = 12.7:1.2:1), which is composed mainly of TiO2, ZrO2, and Nb2O5, with a small amount of TiO and Ti2O3 [30, 41]. Note however that the oxidized surface is strongly depleted in both zirconium and niobium in comparison to the alloy. Sample anodized at 140 V is enriched in Nb with respect to sample anodized at 100 V (Ti/Zr/Nb = 13.1:1:2.3). For both samples, the main component of the surface layer is TiO2 (Fig. 3). The fitted doublet formed by two lines at the binding energies of 459.3 eV (Ti2p3/2) and 465.0 eV (Ti2p1/2) can be ascribed to tetravalent Ti, and the position of the lines corresponds to that of titanium dioxide. Analysis of the XPS lines coming from Nb and Zr indicates to the formation of the well-defined oxidation states related with the formation of Nb2O5 and ZrO2 (Fig. 3). The positions of the lines are higher on the binding energy scale than in bulk oxides by 0.5–0.8 eV. We found this tendency also in the other electrochemically polished samples of the Ti–13Nb–13Zr alloys. It may be related with the formation of nanosize oxide particles within the film or formation of more complex compounds [30]. Oxygen 1s line can be fitted by three components with the relative intensity varying between the samples (Fig. 3). The lowest binding energy peak situated at about 530.4–530.6 eV can be ascribed to TiO2, Nb2O5, and ZrO2. The highest energy one comes from the adsorbed water (533.3 eV) while the middle intensity peak about 531.85 eV can be associated to other compounds containing oxygen, mainly containing phosphorus. The species adsorbed at the surface, as hydroxides for example, can also contribute to this O 1s photoemission line. Phosphorous line is formed by a spin–orbit doublet which is not resolved. In the case of both samples, the position of the main P2p3/2 line can be attributed to phosphates. The N 1s line has two components (XPS data not shown). The higher binding energy line may be attributed to ammonium fluoride which is the component of the electropolishing bath [30]. The structure of the carbon 1s photoemission line is similar for both samples (XPS data nor shown). The dominant line at about 285 eV comes from hydrocarbons adsorbed on the surface.

The deconvoluted spectra of the Ti 2p, Nb 3d, Zr 3d, O 1s, and P 2p core excitations of the Ti–13Nb–13Zr alloy samples anodized at 100 and 140 V
The dependence of the thickness of the prepared oxide layers on the voltage of anodization is presented in Table 2. At lower voltages of anodization, the thickness of the oxide does not exceed 300 nm. The mean increase of the thickness of the oxide layer on the Ti–13Nb–13Zr alloy is 3–4 nm V−1. The increase of the voltage to 140 V causes the increase of the oxide layer thickness to more than 1 μm. In that case, a typical three-layer structure, characteristic for the process of oxidation at the voltage higher than the breakdown voltage, is observed (Fig. 4). The first layer, adhering directly to the substrate, is a barrier-type compact oxide layer, 350-nm thick. Closed pores appear in the second layer, whereas the third layer shows open pores. The growth of an oxide layer via plasma electrolytic oxidation, local breakdown with a small spark, and the subsequent oxygen gas evolution resulted in the formation of this complicated structure [42, 43].

Cross-sectional image of the Ti–13Nb–13Zr alloy sample anodized at 140 V
The EIS is a very useful method for the determination of the corrosion resistance of the Ti–13Nb–13Zr alloy [30, 44-48]. Though Nyquist plots are relatively featureless, the Bode plots are more indicative for the changes in the electrochemical characteristics of the system during aging. The Bode plots of the spectra recorded during first 24 h of the contact of the sample anodized at the voltage of 60 V with the 5 % NaCl solution are presented in Fig. 5.

Impedance spectra of sample anodized at 60 V in body representation: diamonds—just after immersion in 5 % NaCl solution; triangles—when 2 hs elapsed after immersion; squares—when 4 hs elapsed after immersion
The spectra of electropolished samples were previously described in more details elsewhere [30]; in the present paper, they will be invoked only for the sake of comparison. For all anodized samples, the phase angle is more negative than −80° for the whole period of time and almost all frequencies, indicating highly capacitive behavior, typical for passive metals. The plot of the logarithm of modulus of impedance versus logarithm of frequency is practically a straight for all samples and the whole time, but the impedance slightly increases during the period of immersion (Fig. 5). It indicates that the oxide layer is being continuously sealed. For the interpretation of the spectra, we applied the equivalent electrical circuit (EEC) presented in Fig. 6. That EEC consists of the resistance of the electrolyte (R 0) and three sub-circuits connected in series. Each sub-circuit consists of the parallel connection of a resistance (R) and the constant phase element (CPE) which impedance may be expressed by the formula [30]:

The equivalent electrical circuit used for fitting data
where j is the imaginary unit, f is the frequency, f 0 is the frequency of reference, C 0 is the capacitance at the frequency of reference, and α is a coefficient which is close to 1 for an ideal capacitor (in this case, CPE reduces to a capacitor) and assumes usually the value between 0.7 and 1.0 in the case of a nonideal capacitance. The frequency of reference was assumed 1,000 Hz. The equation describing the impedance of the assumed EEC was fitted to the measured data using the program Minuit [49] based on a complex nonlinear regression least-square fitting procedure. The quality of the fit was characterized by the standard deviation s, calculated from the formula:
where Z i means the impedance at the frequency number i and n is the number of frequencies in the impedance spectrum. The quality of fit may be also evaluated visually in Fig. 7, where the values of impedance, calculated by the fitting program, are marked by the empty circles.

The example of the fit to measured data. a Bode plot. b Nyquist plot
The parameter best characterizing corrosion resistance is the polarization resistance at the corrosion potential R pol. For the corrosion process described by the EEC that was used in the present work, it may be calculated from the formula:
The polarization resistances just after the immersion for all samples are given in Table 2. It is to be seen that the sample anodized at the voltage of 100 V shows much better corrosion resistance at all times than any other sample, except the sample anodized at the voltage of 140 V. However, the oxide layer on the latter was much thicker than in the case of the other samples. Note that the polarization resistance of the sample anodized at the voltage of 20 V was lower than in the case of the electropolished sample, which may be ascribed to the imperfect structure of the former.
Conclusions
The Ti–13Nb–13Zr alloy may be effectively covered by the oxide layer composed of the oxides of the metals constituting the alloy in their highest oxidation state, using the method of plasma anodization. The alloy covered with the oxide layer shows very high corrosion resistance. Both the thickness of the oxide layer and the corrosion resistance of the oxide covered alloy increase with the voltage of anodization. Compact oxide layers of the thickness below 1 μm are obtained if the voltage of anodization is not higher than approximately 100 V. Using excessive voltage (140 V) leads to the creation of a very thick layer, showing however relatively high porosity.
References
Geetha M, Singh AK, Asokamani R, Gogia AK (2009) Prog Mater Sci 54:397–425
Evans WJ (1998) Mater Sci Eng A 243:89–96
Niinomi M (1998) Mater Sci Eng A 243:231–236
Kuroda D, Niinomi M, Morinaga M, Kato Y, Yashiro T (1998) Mater Sci Eng A 243:244–249
Ankem S, Greene CA (1999) Mater Sci Eng A 263:127–131
Kohn DH (1998) Curr Opin Solid State Mater Sci 3:309–316
Strzała A, Simka W, Marszałek M (2012) Acta Phys Pol A 121:561–564
Sarró MI, Moreno DA, Ranninger C, King E, Ruiz J (2006) Surf Coat Technol 201:2807–2812
Vasilescu C, Drob P, Vasilescu E, Demetrescu I, Ionita D, Prodana M, Drob SI (2011) Corros Sci 53:992–999
Łaskawiec J, Michalik R (2002) Theoretical and application issues in implants. Silesian University of Technology Publishing House, Gliwice
Wennig R, Kirsch N (1988) In: Seiler HG, Siegel H, Siegel A (eds) Handbook on toxicity of inorganic compounds. Marcel Dekker, New York
Kobayashi E, Wang T, Doi H (1998) J Mater Sci Mater Med 9:567–574
Ramsden JJ, Allen DM, Stephenson DJ, Alcock JR, Peggs GN, Fuller G, Goch G (2007) Annal CIRP 56:687–711
Chen ZX, Wang WX, Takao Y, Matsubara T, Ren LM (2011) Appl Surf Sci 257:7254–7262
Jacobs JJ, Skipor AK, Black J, Urban R, Galante JO (1991) J Bone Joint Surg 73:1475–1486
Ku CH, Pioletti DP, Browne M, Gregson PJ (2002) Biomaterials 23:1447–1454
Davidson JA, Mishra AK, Kovasc P, Poggie RA (1994) Biomed Mater Eng 4:231–243
Davidson JA, Kovacs P (1992) USA Patent no. 4.169.597
Wang K (1996) Mater Sci Eng A 213:134–137
Szewczenko J, Jaglarz J, Basiaga M, Kurzyk J, Paszenda Z (2013) Opt Appl 43:173–180
Deplancke JL, Garnier A, Massiani Y, Winand R (1994) Electrochim Acta 39:1281–1289
Shibata T, Zhu YC (1995) Corros Sci 37:253–270
Krząkała A, Służalska K, Dercz G, Maciej A, Kazek A, Szade J, Winiarski A, Dudek M, Michalska J, Tylko G, Osyczka AM, Simka W (2013) Electrochim Acta 104:425–438
Krząkała A, Służalska K, Widziołek M, Szade J, Winiarski A, Dercz G, Kazek A, Tylko G, Michalska J, Iwaniak A, Osyczka AM, Simka W (2013) Electrochim Acta 104:407–424
Song HJ, Kim MK, Jung GC, Wang MS, Park YJ (2007) Surf Coat Technol 201:8738–8745
Niemeyer TC, Grandinia CR, Pinto LMC, Angelo ACD, Schneider SG (2009) J Alloy Comp 476:172–175
Durgalakshmi D, Chandran M, Manivasagam G, Rao MSR, Asokamani R (2013) Tribol Int 63:132–140
Cvijović-Alagić I, Cvijović Z, Mitrović S, Panić V, Rakin M (2011) Corros Sci 53:796–808
Kumar P, Babu PD, Mohan L, Anandan C, Grips VKW (2013) J Mater Eng Perform 22:283–293
Simka W, Mosiałek M, Nawrat G, Nowak P, Żak J, Szade J, Winiarski A, Maciej A, Szyk-Warszyńska L (2012) Surf Coat Technol 213:239–246
Saji VS, Choe HC, Brantley WA (2009) J Mater Sci 44:3975–3982
Saji VS, Choe HC (2009) Corros Sci 51:1658–1663
Ossowska A, Zieliński A, Supernak M (2012) Solid State Phenom 183:137–142
Duarte LT, Biaggio SR, Rocha-Filho RC, Bocchi N (2011) J Mater Sci Mater Med 22:1663–1670
Duarte LT, Biaggio SR, Rocha-Filho RC, Bocchi N (2013) Corros Sci 72:35–40
Simka W (2011) Electrochim Acta 56:9831–9837
Simka W, Krząkała A, Masełbas M, Dercz G, Szade J, Winiarski A, Michalska J (2013) RSC Adv 3:11195–11204
Simka W, Socha RP, Dercz G, Michalska J, Maciej A, Krząkała A (2013) Appl Surf Sci 279:317–323
Oliveira NTC, Ferreira EA, Duarte LT, Biaggio SR, Rocha-Filho RC, Bocchi N (2006) Electrochima Acta 51:2068–2075
Oliveira NTC, Biaggio SR, Nascente PAP, Rocha-Filho RC, Bocchi N (2006) Surf Interface Anal 38:410–412
Baker MA, Assis SL, Higa OZ, Costa I (2009) Acta Biomater 5:63–75
Ha JY, Tsutsumi Y, Doi H, Nomura N, Kim KH, Hanawa T (2011) Surf Coat Technol 205:4948–4955
Xue W, Zhu Q, Jin Q, Hua M (2010) Mater Chem Phys 120:656–660
Assis SL, Wolynec S, Costa I (2006) Electrochim Acta 51:1815–1819
Shukla AK, Balasubramaniam R (2006) Corros Sci 48:1696–1720
Assis SL, Rogero SO, Antunes RA, Padilha AF, Costa I (2005) J Biomed Mater Res B 73B:109–116
Robin A, Carvalho OAS, Schneider SG, Schneider S (2008) Mater Corros 59:929–933
Assis SL, Costa I (2007) Mater Corros 58:329–333
James F, Roos M (1975) Comput Phys Commun 10:343–367
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Mosiałek, M., Nawrat, G., Szyk-Warszyńska, L. et al. Anodic oxidation of the Ti–13Nb–13Zr alloy. J Solid State Electrochem 18, 3073–3080 (2014). https://doi.org/10.1007/s10008-014-2446-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-014-2446-7