
Abstract
The main goal of this work was to obtain a calculated absorption spectrum of rhodamine 800 in an aqueous solution, which most accurately reproduces the experimental one. To achieve this result, I used the hybrid functionals supported by Gaussian 16 software package. In this case, the basis set (6-31++G(d,p)) and the solvent model (IEFPCM) were not varied. The B3PW91 functional gave the best agreement with the experimental absorption spectrum of the dye in an aqueous medium. B3P86, B971, B972, B98, X3LYP, APF, HSE06, and N12SX functionals also give good absorption energy coincidence. The B3PW91/6-31++G(d,p)/IEFPCM theory level chosen in this way made it possible to calculate the various characteristics of rhodamine 800 in the ground and excited states. An important result of this work was the establishment of the vibronic nature of the short-wavelength smaller maximum of the absorption spectrum. The influence of the strong H-bond of the exocyclic nitrogen atom with the water molecule on the dye excitation was analyzed.
Graphical abstract

Similar content being viewed by others


Data availability
All additional data is contained in the attachment.
Code availability
Not applicable.
References
Rahavendran SV, Karnes HT (1996) Application of Rhodamine 800 for reversed phase liquid chromatographic detection using visible diode laser-induced fluorescence. Anal Chem 68:3763–3768
Abugo OO, Nair R, Lakowicz JR (2000) Fluorescence properties of Rhodamine 800 in whole blood and plasma. Anal Biochem 279:142–150
Sakanoue J, Ichikawa K, Nomura Y, Tamura M (1997) Rhodamine 800 as a probe of energization of cells and tissues in the near-infrared region: a study with isolated rat liver mitochondria and hepatocytes. J Biochem 121:29–37
Imasaka T, Tsukamoto A, Ishibashi N (1989) Visible semiconductor laser fluorometry. Anal Chem 61:2285–2288
Mayer GV, Kopylova TN, Andreev YM, Svetlichnyi VA, Telminov EN (2009) Parametrical conversion of the frequency of organic lasers into the middle-IR range of the spectrum. Russ Phys J 52:640–645
Iwuji C, Ghann W, Iwuji O, Uddin J (2018) Rhodamine 800 as a sensitizer for dye sensitized solar cells. Nanosci J 1:1–4
Bakker RM, Drachev VP, Liu Z, Yuan H-K, Pedersen RH, Boltasseva A, Chen J, Irudayaraj J, Kildishev AV, Shalaev VM (2008) Nanoantenna array-induced fluorescence enhancement and reduced lifetimes. New J Phys 10:125022
Valmorra F, Bröll M, Schwaiger S, Welzel N, Heitmann D, Mendach S (2011) Strong coupling between surface plasmon polariton and laser dye rhodamine 800. Appl Phys Lett 99:051110
K. Sekiguchi, S. Yamaguchi, T. Tahara, Formation and dissociation of Rhodamine 800 dimers in water: steady-state and ultrafast spectroscopic study, J Phys Chem A 110 (2006) 2601–2606.
Sanchez-Valencia JR, Aparicio FJ, Espinos JP, Gonzalez-Elipe AR, Barranco A (2011) Rhodamine 6G and 800 J-heteroaggregates with enhanced acceptor luminescence (HEAL) adsorbed in transparent SiO 2 GLAD thin films. Phys Chem Chem Phys 13:7071–7082
Suklabaidya S, Chakraborty S, Saha J, Dey B, Sarkar S, Bhattacharjee D, Hussain SA (2021) Study of polydiacetylenes and rhodamine-800 mixed flm at air–water interface and onto solid support: trace of fuorescence resonance energy transfer (FRET). Polym Bull 78:93–113
Menzel R, Bornemann R, Thiel E (1999) Influence of chemical substitution and electronic effects on the triplet state kinetics of xanthene dyes. Phys Chem Chem Phys 1:2435–2440
Sekiguchi K, Yamaguchi S, Tahara T (2008) Femtosecond time-resolved electronic sum-frequency generation spectroscopy: a new method to investigate ultrafast dynamics at liquid interfaces. J Chem Phys 128:114715
Werner JPF, Huang Y, Mishra K, Janowski R, Vetschera P, Heichler C, Chmyrov A, Neufert C, Niessing D, Ntziachristos V, Stiel AC (2020) Challenging a preconception: optoacoustic spectrum differs from the optical absorption spectrum of proteins and dyes for molecular imaging. Anal Chem 92:10717–10724
Alessi A, Salvalaggio M, Ruzzon G (2013) Rhodamine 800 as reference substance for fluorescence quantum yield measurements in deep red emission range. J Luminesc 134:385–389
Le KQ, Dang NH (2018) Photoluminescence spectroscopy of Rhodamine 800 aqueous solution and dye-doped polymer thin-film: concentration and solvent effects. J Electron Mater 47:4813–4817
Zhou P (2018) Why the lowest electronic excitations of rhodamines are overestimated by time-dependent density functional theory, Int J Quantum Chem e25780
Cossi M, Rega N, Scalmani G, Barone V (2003) Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comp Chem 24:669–681
Adamo C, Jacquemin D (2013) The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem Soc Rev 42:845–856
Charaf-Eddin A, Planchat A, Mennucci B, Adamo C, Jacquemin D (2013) Choosing a functional for computing absorption and fluorescence band shapes with TD-DFT. J Chem Theory Comput 9:2749–2760
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16, Revision C.01, Inc., Wallingford CT
Condon EU (1928) Nuclear motions associated with electron transitions in diatomic molecules. Phys Rev 32:858–872
Improta R, Barone V, Scalmani G, Frisch MJ (2006) A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution. J Chem Phys 125:054103
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3093
Scalmani G, Frisch MJ, Mennucci B, Tomasi J, Cammi R, Barone V (2006) Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J Chem Phys 124:094107
Baiardi A, Bloino J, Barone V (2013) General time dependent approach to vibronic spectroscopy including Franck-Condon, Herzberg-Teller, and Duschinsky Effects. J Chem Theory Comput 9:4097–4115
Herzberg G, Teller E (1933) Schwingungsstruktur der Elektronenubergange bei mehratomigen Molekulen, Z. Phys Chem Abt B 21:410–446
Santoro F, Lami A, Improta R, Bloino J, Barone V (2008) Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg-Teller effect: The Qx band of porphyrin as a case study. J Chem Phys 128:224311
Duschinsky F (1937) The importance of the electron spectrum in multi atomic molecules. Concerning the Franck-Condon principle. Acta Physicochim URSS 7:551
Charaf-Eddin A, Planchat A, Mennucci B, Adamo C, Jacquemin D (2013) Choosing a functional for computing absorption and fluorescence band shapes with TD-DFT. J Chem Theory Comput 9:2749–2760
Alia JD, Flack JA (2020) Unspecified verticality of Franck-Condon transitions, absorption and emission spectra of cyanine dyes, and a classically inspired approximation. RSC Adv 10:43153–43167
Jacquemin D, Adamo C (2012) Basis set and functional effects on excited-state properties: three bicyclic chromogens as working examples. Int J Quantum Chem 112:2135–2141
Dennington R, Keith TA, Millam JM (2016) GaussView, Version 6.1, Semichem Inc., Shawnee Mission KS
Jacquemin D, Brémond E, Ciofini I, Adamo C (2012) Impact of vibronic couplings on perceived colors: two anthraquinones as a working example. J Phys Chem Lett 3:468–471
Dierksen M, Grimme S (2004) The vibronic structure of electronic absorption spectra of large molecules: a time-dependent density functional study on the influence of “exact” hartree-fock exchange. J Phys Chem A 108:10225–10237
Kantchev EAB, Norsten TB, Sullivan MB (2012) Time-dependent density functional theory (TDDFT) modelling of Pechmann dyes: from accurate absorption maximum prediction to virtual dye screening. Org Biomol Chem 10:6682–6692
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Burke K, Perdew JP, Wang Y (1998) Derivation of a generalized gradient approximation: the PW91 density functional. In: Dobson JF, Vignale G, Das MP (eds) Electronic density functional theory. Springer, Boston
Reichardt C (1994) Solvatochromic dyes as solvent polarity indicators. Chem Rev 94:2319–2358
Singh UC, Kollman PA (1984) An approach to computing electrostatic charges for molecules. J Comput Chem 5:129–145
Zhao G-J, Han K-L (2012) Hydrogen bonding in the electronic excited state. Acc Chem Res 45:404–413
Sierański T (2017) Discovering the stacking landscape of a pyridine-pyridine system. J Mol Model 23:338
Cerny J, Hobza P (2005) The X3LYP extended density functional accurately describes H-bonding but fails completely for stacking. Phys Chem Chem Phys 7:1624–1626
Lenzi V, Driest PJ, Dijkstra DJ, Ramos MMD, Marques LSA (2019) Investigation on the intermolecular interactions in aliphatic isocyanurate liquids: revealing the importance of dispersion. J Mol Liquids 280:25–33
Austin A, Petersson GA, Frisch MJ, Dobek FJ, Scalmani G, Throssell K (2012) A Density functional with spherical atom dispersion terms. J Chem Theory Comput 8:4989–5007
Author information
Authors and Affiliations
Contributions
The author confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results, and manuscript preparation.
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 6171 kb)
Appendix. R800 dimer
Appendix. R800 dimer
As mentioned above, the main absorption maximum of the R800 dimer coincides with the short-wavelength vibronic absorption peak of the dimer (see Fig. 4). This interesting feature prompts the calculation of absorbance for the dye dimer as well. Detailed structural information about it is absent in the literature. Only in Ref. [9], it was suggested that this should be a non-parallel H-dimer. Therefore, in this work, its structure was chosen for reasons of steric correspondence and geometry optimization by minimizing the potential energy. First of all, it should be noted that the mutual steric constraints of the hydrogen atoms of the aliphatic rings exclude the parallel (ring over the ring) arrangement of the monomers. The cruciform structure of the dimeric complex turned out to be stable (Fig. 11).

Optimized structure of the R800 dimer. Cartesian coordinate axes are directed along principal axes of inertia

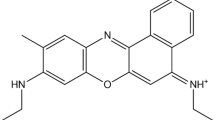
Molecular structure of rhodamine 800

Diagram of vibronic absorption transition

Calculated maxima Evibron (eV) of R800 vibronic absorption spectra in aqueous solution. The functionals are arranged in ascending order of the X percentage of exact Hartree-Fock exchange in them (X values are indicated in parentheses). For functionals with long-range correction, X values for small and large distances are indicated, for the HISS functional — for small, medium, and long distances, respectively. The bold horizontal line is the experimental maximum of the R800 dilute aqueous solution (λmax = 690 nm, Emax = 1.80 eV [9])

The calculated vibronic absorption spectrum of R800 in aqueous media (thin line) and the corresponding experimental spectrum (1.3 μM) (thick line, adapted with permission from Ref. [9]). Inset: spectrophotometrically resolved monomer (solid line) and dimer (dashed line) spectra adapted with permission from Ref. [9]. Copyright 2006 American Chemical Society. The vertical sticks are the dipole strength of the vibronic transitions from Table S2 (see Supplementary Material)

Calculated IR spectra of R800 in an aqueous media. The vibration frequencies involved in vibronic transitions (see Table S2 in Supplementary Material) are indicated by red arrows

HOMO (left) and LUMO (right). Positive lobes are shown in red and negative lobes in blue

The electron density difference between Franck-Condon point and the ground state of R800 cation in an aqueous media. Regions of positive values are shown in red and negative values in blue

Structure of the R800 hydrated complex. A strong hydrogen bond is shown by a dotted line. Its length in Å (distance between heavy atoms) is given for the ground and equilibrium excited (in parentheses) states

Calculated vibronic absorption spectrum of the “R800+H2O” complex (thin line) and the experimental spectrum (1.3 μM of R800 in water) from Ref. [9] (thick line)

The electron density difference between Franck-Condon point and the ground state of the “R800+H2O” system in an aqueous media
In addition, optimization of the geometry using the B3PW91 functional, which gave the best coincidence of the calculated spectrum of the monomer with the experimental one (see above), led to its destruction. This result is not unexpected, since the correct accounting of dispersion interactions requires the use of specialized functionals [42,43,44]. From the set of author’s hybrid functionals supported by Gaussian16 and including dispersion, the APFD [45] was chosen, since it gave the absorption energy for the monomer closest to the experiment compared with PW6B95D3 and ωB97XD (see Fig. 3). Indeed, optimization at the APFD/6-31++G(d,p)/IEFPCM theory level gave a stable structure of a dimeric complex with a distance between chromophores of 3.3 Å (see Fig. 11). However, in the IR spectrum of vibrations of the ground state of the dimeric complex (Fig. S11), one imaginary frequency was still present (synchronous vibrations of C3 and C22 atoms perpendicular to the chromophore plane, see Supplementary Material). This prevented the calculation of vibronic transitions, as was done for the dye monomer. The optimization of the excited state of the R800 dimer due to computational complexity could not be performed in a reasonable time using the available computing power. Therefore, we will be content with the analysis of vertical transitions in the dimer (Table 3). For correct comparative analysis, Table 3 also shows the corresponding data for the monomer, calculated at the same theory level.
It can be seen from it that the three lowest absorption transitions are due to the same four transitions between MOs participating in each of them in different ratios. Of these, only the S0 → S3 transition has a significant oscillator strength f; therefore, the main absorption peak of the dimer corresponds to it (see the inset in Fig. 4). From Table 3, it can be seen that the S0 → S3 excitation energy of the dimer (2.14 eV) is very close to the S0 → S1 excitation energy of the monomer (2.17 eV). This almost equality of the excitation energies of the monomer and dimer, found experimentally in Ref. [9], prompted its authors to conclude that the S1 dimer dissociates to produce the S1 and S0 monomers or relaxes to the S0 dimer. However, the theoretical calculation performed in the present work refined this result: the excitation of the dimer is due to the electronic S0 → S3 transition.
Rights and permissions
About this article

Cite this article
Kostjukov, V.V. Excitation of rhodamine 800 in aqueous media: a theoretical investigation. J Mol Model 28, 52 (2022). https://doi.org/10.1007/s00894-022-05034-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05034-w