
Abstract
Radix puerariae—a popular traditional Chinese medicine—is used for the treatment of diarrhea, acute dysentery, deafness, and cardiovascular diseases. It can also be used as an effective antioxidant and has been tested as an anticancer drug. Daidzein and puerarin are its main active compounds. The present contribution was focused on experimental and theoretical studies of the 1H and 13C NMR chemical shifts and nuclear magnetic shielding parameters of daidzein and puerarin. Experimental data were gained by exploring standard one-dimensional spectra and a set of two-dimensional measurements: COSY, HSQC, and HMBC. The theoretical gauge independent atomic orbital density functional theory supporting studies, were performed to determine shielding constants and chemical shifts in vacuum and methanol-d4 solvent. The correlation between experimental and theoretical data was fairly good, especially when the DFT/PBE0 approach was used. The molecular properties of daidzein and puerine related to antiradical activity were studied in the context of a single-step hydrogen atom transfer mechanism and its correlation with 13C NMR chemical shifts.

Electron spin density distribution for daidzein radical
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In China, traditional Chinese medicine (TCM) based on natural products plays an important role, and accounts for approximately 40% of total consumption of pharmaceuticals. However, the active components of plants and their pharmacological mechanisms are still not well recognized and understood. Chinese scholars believe that the curative effect of some herbal medicines is closely related to the antioxidative activity found in flavonoids, saponins, polysaccharides, organic acids, terpenes, etc. [1].
Radix puerariae, belonging to the families of fabaceae or leguminosae, has been used traditionally for the treatment of diarrhea, acute dysentery, deafness, and cardiovascular diseases. Since 1950, interest in its phytochemistry and pharmacological activity has increased significantly [2, 3]. Over 70 chemical constituents of radix puerariae have been identified. This collection includes daidzein (Fig. 1a) and puerarin (Fig. 1b), daidzin, genistein, genistin, and formononetin. Puerarin—the first identified major isoflavone—constitutes the greatest part [1.88–2.55% (w/w)] of this plant [4]. Puerarin, daidzin, and daidzein have been reported to exhibit estrogenic, antiestrogenic, and anti-inflammatory activities [5, 6]. Studies indicate that the cardioprotective effects of puerarin against ischemia and reperfusion injury are mediated by opening the calcium-activated potassium channels and activating protein kinase C—both effects may be due to an antioxidative activity [7]. Also genistein, being an isoflavone, possesses antioxidative and pharmacologic activities against cardiocerebrovascular diseases; its activity is based on scavenging of oxygen free radicals [8, 9].

Chemical schemes and structures of a daidzein and b puerarin (daizein-8-C-glucoside)
To date, several pharmacological studies on puerarin, daidzin have been performed, applying liquid chromatography (LC), joint liquid chromatography with mass spectrometry (LC–MS), and liquid chromatography–electrospray ionization–mass spectrometry (LC-ESI-MS) to provide chemical properties of investigated compounds [10,11,12]. NMR chemical shifts of daidzein and puerarin have also been measured [13,14,15]; however, magnetic shielding has never been reported for either compound. The present work reports experimental 1H and 13C NMR chemical shifts of daidzein and puerarin dissolved in methanol-d4. For the same solutions, we also completed measurements of magnetic shielding using an original method from our laboratory [16]. By considering features of 13C NMR spectroscopy, chemical shifts can be used as promising descriptors for quantitative structure–activity relationships [17].
Daidzein [7-hydroxy-3-(4-hydroxyphenyl) chromen-4-one] and puerarin (the 8-C-glucoside of daidzein) are isoflavones [18, 19]. Molecular properties related to their antioxidative activity are presented. The 1H and 13C NMR measurements of nuclear magnetic shielding and chemical shifts were determined, and supplementary theoretical studies allowed for its careful analysis. Theoretical studies include molecular and corresponding radical structures, atomic charge distribution, electron spin density distribution for radicals, and O–H bond dissociation energies (BDE).
Methods
Experimental
The NMR spectra of daidzein and puerarin were obtained at 298.2 K using a Bruker AVANCE III HD 500 MHz spectrometer operating at 500.20 and 125.79 MHz for 1H and 13C nuclei, respectively, with the 5 mm CPPBBO BB probehead and 5 mm o.d. glass tubes (528PP, Wilmad, https://www.wilmad-labglass.com). Liquid CD3OD was used as the reference of shielding and lock solvent at the same time. 1H and 13C shielding values were determined relative to the CD3− signal of pure methanol-d4. All 1H and 13C shielding values were obtained on the basis of observed absolute resonance frequencies of studied samples. The 1H and 13C NMR chemical shifts were measured relative to pure liquid TMS used as external reference standards [20, 21]. The assignment of chemical shifts to individual atoms was completed on the above basis and additionally using the variety of two-dimensional(2D: 1H-1H COSY, 1H-13C HSQC and 1H-13C HMBC) spectra, shown as Figs. S1–S10 in the Supporting Materials.
Computations
All calculations were performed within the density functional theory (DFT) [22], applying B3LYP [23,24,25], M06-2X [26], and PBE0 [27] functionals with standard aug-cc-pVTZ [28] and 6-311G(2d, 2p) atomic basis sets [29]. The structure of molecules was optimized for every combination of functional/basis set, and frequency calculations additionally confirmed the proper determination of equilibrium geometries. Enthalpy values were calculated for normal conditions. The resulting structures were used for further studies. The gauge independent atomic orbital (GIAO) method was used for carrying out magnetic shielding parameters [30,31,32], and theoretical chemical shifts were determined by using the corresponding calculations for tetramethylsilane (TMS) (Table 1). The polarized continuum model (PCM) was applied to represent solvent [33]. All calculations were performed utilizing Gaussian16 code [34]. Electron spin density plots were produced by applying the GausView6 program [35].
Results and discussion
Experimental and theoretical NMR studies
We used the same experimental and theoretical procedures in our present study for daidzein and puerarin as in a previous report [36]. We performed a complete set of 1H and 13C frequency measurements for chemicals dissolved in methanol-d4. According to Equations (1) and (2) [16], the shielding isotropic parameters of protons and carbon nuclei (σH and σC) were determined as follows:
where νH, νC, and νD are the resonance frequencies of protons, carbons, and deuterons, respectively. The resonance frequency of –CD3− deuterons in methanol-d4 was νD = 76.7839977 MHz, and the deuteron shielding constant was σD* = 0.000029593 [16]. Note that the application of Eqs. 1 and 2 requires the use of real values of shielding, not expressed in ppm.
The analogous characterization was performed theoretically. A comparison of new experimental and calculated results is shown in Table 2. Here, we present experimental and calculated magnetic shielding parameters of daidzein. The theoretical results are given for an isolated molecule (a molecule in vacuum) and for the same molecule dissolved in methanol-d4. According to root mean square (RMS) values, the correlation between calculated and experimental shielding parameters in methanol-d4 is better than similar calculations for isolated molecules. For example, the RMS value in methanol-d4 (7.41) calculated by the PBE0/aug-cc-pVTZ approach is lower than the value 7.63 for the molecule in vacuum, and values used B3LYP/aug-cc-pVTZ and M06-2X/aug-cc-pVTZ are 7.54 and 8.77 in methanol-d4, which are also lower than values (7.58 and 9.10) in vacuum. For different functionals within the same basis set, the PBE0 functional is better comparing to B3LYP and M06-2X in methanol-d4.
The 13C and 1H chemical shifts of daidzein determined experimentally and theoretically are presented in Table 3. Comparing the correlation between experimental and calculated data, PBE0 is again better than the other two functionals. RMS values for B3LYP and PBE0 with the same basis set are also better in methanol-d4 than for the isolated molecule. The RMS value of chemical shift in methanol-d4 calculated in the PBE0/aug-cc-pVTZ approach is 7.51, which is lower than the value of 7.69 for the molecule in vacuum. Using B3LYP/aug-cc-pVTZ approach, values are 7.63 in methanol-d4 and 7.82 in vacuum. For chemical shifts, the basis set 6-311G(2d,2p) is better than aug-cc-pVTZ. RMS values are 7.38 for the PBE0/6-311G(2d,2p) approach in methanol-d4 and 7.51 for PBE0/aug-cc-pVTZ, and B3LYP and M06-2X methods present same conclusion, i.e., that the basis set 6-311G(2d,2p) is better than aug-cc-pVTZ both in methanol-d4 and in vacuum. Obviously, the differences of experimental and theoretical 1H chemical shifts are small (almost within ±1 ppm), showing high consistency between experimental and calculated results. The correlation between experimental and theoretical chemical shifts (δ) for daidzein is shown in Fig. 2b. This correlation looks better than the previous plot shown for shielding in Fig. 2a. In particular, the plot crosses the origin of coordinates precisely in Fig. 2b.

Tables 4 and 5 present the 13C and 1H shielding values and chemical shifts of puerarin determined experimentally and theoretically in the two phases as before. The differences in 13C shielding constants for atoms C4, C6, C8, C3’ and C5’ are much higher in our list (from −16.07 ppm to 40.84 ppm for PBE0/aug-cc-pVTZ method in vacuum)—the correlation line is shown in Fig. 3a. However, 1H shielding changes are similar to those of daidzein between −4.48 ppm [H(6)] and −6.37 ppm [H(5)]. Results in Table 4 in methanol-d4 are much better than in vacuum for the same method PBE0/aug-cc-pVTZ, with values ranging from −18.74 ppm to 38.82 ppm. In Table 5, the PBE0 method also presents better RMS results compared to the other method, RMS values in methanol-d4 are 7.67 for PBE0/aug-cc-pVTZ and 7.55 for PBE0/6-311G(2d,2p). It seems that our experimental data for chemical shifts compared to results of Xu et al. [14] match our calculated values well. For example, differences between experimental data and calculated values with PBE0/aug-cc-pVTZ method methanol-d4 are smaller than differences between data of of Xu et al. [14] and our calculated values (only values C(2′), C(3′), C(5′), and C(6′) are higher).

Comparing Tables 3 and 5, experimental data of shielding changes between daidzein and puerarin for each atom are quite small, ranging from −1.52 ppm [C(3′)] to 1.51 ppm [C(2′)] based on their similar chemical structure. However, RMS values of puerarin present better results than daidzein with every method used. For instance, the RMS value of puerarin is 5.73 ppm for PBE0/aug-cc-pVTZ in methanol-d4 with 1.68 less than daidzein, and RMS differences of shielding between daidzein and puerarin ranging from 0.93 ppm [M06-2X/6-311G(2d,2p) in methanol-d4)] to 2.47 ppm (B3LYP/aug-cc-pVTZ in methanol-d4). Differences of chemical shifts in Tables 4 and 6 from experimental results are a little higher than shielding comparing the same atom between daidzein and puerarin.
Antioxdative properties
The optimization of daidzein and puerarin structures leads to a nonplanar flovone backbone with a dihedral angle between benzopyrene and phenyl moieties of about 40° (Table 6). The corresponding radicals, resulting from hydrogen abstraction from phenolic groups, are only slightly more planar. The transformation of −OH into =O• substituent results in a shorter C–O bond with a significant double bond character. The effect, however, is not strong enough to flatten the flavone skeleton. Puerarin, which is daidzien modified by a glucoside group, possesses similar structural characteristics. The charge extraction from the flovone ring is too small to cause significant geometrical effects (Table 6).
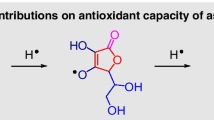
The scavenging of free OH• or OOH• radicals of antioxidants is controlled by their ability to transfer hydrogen of phenolic groups attached directly to aromatic rings according to the following reactions
In the case of the studied compounds, the mechanism of the single-step hydrogen atom transfer dominates other mechanisms involving electron transfer. The thermodynamics of such processes is related directly to the delocalization of unpaired electron on aromatic moieties. The structures of the studied radicals possess significantly broken planarity, restricting the space for electron delocalization.
The mesomeric structures for daidzein rationalize the significant electron density delocalization (Fig. 4). This observation agrees well with the electron spin density distribution (Fig. 5, Fig. S11). Interestingly, some transfer of electron spin density is possible from the phenyl to the benzopyrene fragment. Of the many molecular properties, such as ionization energy, chemical hardness, or spin delocalization influencing the radical scaverging mechanism, the bond dissociation enthalpy (BDE), defined by the reaction
is probably the most important. The information provided by reaction (5) is equivalent to that carried out by (3) or (4). The enthalpy of cleaving the O–H bond by homolysis at a specific temperature is defined as

Possible mesomeric structures of daidzein radical resulting from H dissociation at position C7-OH

Electron spin density distribution for daidzein radical phenolic hydrogen loss on oxygen at a C7 (benzopyrene ring) and b C4’ (phenyl ring)
The reference BDE for OH• or OOH• is adopted as
and the enthalpy of the resulting single-step hydrogen atom transfer (HAT) process is
The O–H BDEs are presented in Table 7. The hydrogen-donating capacities of daidzein and puerarin were calculated for positions (C)-OH and (4′)-OH. The results, when compared to reference OOH• and OH• radicals, indicate the increasing BDE upon hydrogen abstraction from the benzopyrene ring at position C7. Despite the fact that the actual mechanism of action of antioxidants is much more complicated, the HAT parameter constitutes the most important factor for the assessment of oxidative properties. The transition states (Fig. S12) for the hydrogen transfer in HAT favor the C4’ position for the reaction (Table 7). Comparison of 13C chemical shifts, in agreement with other suggestions [17], indicates a correlation with BDE (and HAT) (Fig. S13).
Conclusions
Daidzein and puerarin represent two active compounds of Radix puerariae—an important member of the TCM collection. The medicinal activity of these compounds is believed to be related to their free radical scavenging activity. Puerarin and daidzein are isoflavones and are well known for their cardioprotective effects. These two chemicals were studied experimentally, providing a complete NMR analysis and determining the full set of 1H and 13C chemical shifts and corresponding shielding constants. The measurements were supported by theoretical considerations within DFT methodology. The correlation between calculated and experimental shielding parameters and chemical shifts is generally good, especially when the PBE0 approach is used. The antioxidative properties of daidzein and puerarin were studied with reference to hydroxyl and hydroperoxyl radicals. The calculated BDEs indicate the promising antiradical properties of these compounds. The single-electron hydrogen atom transfer molecular mechanism seems to be a suitable way to activate these molecules. The observed correlation between 13C chemcial shifts and BDE indicates that NMR measurements may provide useful data for molecules of interest.
References
Li QH, Li TL, Huang LL, Li F (2008) Research progress on antioxidant activities and evaluation of Chinese medicine. Lishizhen Med Mate Med Res 19:1257
Shibata S, Murakami T, Nishikawa Y, Harada M (1959) The constituents of pueraria root. Chem Pharm Bull 7:134–136
Zhang Z, Lam T-N, Zou Z (2013) Radix puerariae: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J Clin Pharmacol 53:787–811
Wong KH, Li GQ, Li KM, Razmovski-Naumovski V, Chan K (2011) Kudzu root: traditional uses and potential medicinal benefits in diabetes and cardiovascular diseases. J Ethnopharmacol 134:584–607
Overstreet DH, Kralic JE, Morrow AL, Ma ZZ, Zhang YW, Lee DY (2003) NPI-031G (puerarin) reduces anxiogenic effects of alcohol withdrawal or benzodiazepine inverse or 5-HT2C agonists. Pharmacol Biochem Behav 75:619–625
Hu WZ, Yang XJ, Zhe C, Zhang Q, Sun L, Cao KJ (2011) Puerarin inhibits iNOS, COX-2 and CRP expression via suppression of NF-B activation in LPS-induced RAW264.7 macrophage cells. Pharmacol Rep 63:781–789
Sun LH, Hu YM, Zou DL, Ji SG (2000) Determination of puerarin in radix puerariae and naodesheng tablets by HPCE. Zhongguo Yaoxue Zazhi 35:694–696
Gao Q, Yang B, Ye ZG, Wang J, Bruce IC, Xia Q (2007) Opening the calcium-activated potassium channel participates in the cardioprotective effect of puerarin. Euro J Pharm 574:179–184
Han RM, Tian YX, Liu Y, Chen CH, Ai XC, Zhang JP, Skibsted LH (2009) Comparison of flavonoids and isoflavonoids as antioxidants. J Agric Food Chem 57:3780–3785
Park EK, Shin J, Bae EA, Lee YC, Kim DH (2006) Intestinal bacteria activate estrogenic effect of main constituents puerarin and daidzin of Pueraria thunbergiana. Biol Pharm Bull 29:2432–2435
Yan Y, Chai CZ, Wang DW, Wu J, Xiao HH, Huo LX, Zhu DN, Yu BY (2014) Simultaneous determination of puerarin, daidzin, daidzein, paeoniflorin, albiflorin, liquiritin and liquiritigenin in rat plasma and its application to a pharmacokinetic study of ge-gen decoction by a liquid chromatography–electrospray ionization-tandem mass spectrometry. J Pharm Biomed Anal 95:76–84
Yu ZG, Gao XX, Yuan HX, Liu T, Ma MY, Chen XH, Bi KS (2007) Simultaneous determination of safflor yellow A, puerarin, daidzein, ginsenosides (Rg1, Rb1, Rd), and notoginsenoside R1in rat plasma by liquid chromatography–mass spectrometry. J Pharm Biomed Anal 45:327–336
Tanaka K, Ohgo Y, Katayanagi Y, Yasui K, Hiramoto S, Ikemoto H, Nakata Y, Miyoshi N, Isemura MN, Ohashi S (2014) Imai, anti-inflammatory effects of green soybean extract irradiated with visible light. Sci Rep 4:4732
Xu SS, Wang W, He TY (2016) Separation and purification of salidroside, puerarin and epimedium glycoside by high-speed counter current chromatography. J Food Safe Qual 7:4323–4328
Zhao R, Tan T, Sandström C (2011) NMR studies on puerarin and its interaction with beta-cyclodextrin. J Biol Phys 37:387–400
Jackowski K, Jaszunski M, Wilczek M (2010) Alternative approach to the standardization of NMR spectra. Direct measurements of nuclear magnetic shielding in molecules. J Phys Chem A 114:2471–2475
Verma RP, Hansch C (2011) Use of 13C NMR chemical shift as QSAR/QSPR descriptor. Chem Rev 111:2865–2899
Gambini J (2010) Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim Biophys Acta 1802:11–205
Yeung DK, Leung SW, Xu YC, Vanhoutte PM, Man RY (2006) Puerarin, an isoflavonoid derived from radix puerariae, potentiates endothelium-independent relaxation via the cyclic AMP pathway in porcine coronary artery. Eur J Pharm 552:105–111
Harris RK, Becker ED, Cabral de Menezes SM, Goodfellow R, Granger P (2002) NMR nomenclature: nuclear spin properties and conventions for chemical shifts. IUPAC recommendations 2001. Magn Reson Chem 40:489–505
Harris RK, Becker ED, Cabral de Menezes SM, Granger P, Hoffman RE, Zilm KW (2008) Further conventions for NMR shielding and chemical shifts (IUPAC recommendations 2008). Magn Reson Chem 46:582–598
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136:864–871
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Miehlich B, Savin A, Stoll H, Preuss H (1989) Results obtained with the correlation-energy density functionals of Becke and Lee, Yang and Parr. Chem Phys Lett 157:200–206
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120:215–241
Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110:6158–6169
Kendall RA, Dunning Jr TH, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796–6806
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Wolinski K, Hinton JF, Pulay P (1990) Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J Am Chem Soc 112:8251–8260
Ditchfield R (1974) Self-consistent perturbation theory of diamagnetism. Mol Phys 27:789–807
London F (1937) Théorie quantique des courants interatomiques dans les combinaisons aromatiques. J Phys Radium 8:397–409
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3093
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2018) Gaussian 16, Revision E.01, Inc., Wallingford CT
Dennington R, Keith TA, Millam J M (2016) GaussView, Version 6
Yi Y, Adrjan B, Wlodarz J, Li J, Jackowski K, Roszak S (2018) NMR measurements and DFT studies of nuclear magnetic shielding in emodin and chuanxiongzine molecules. J Mol Struct 1166:304–310
Acknowledgments
We thank the Wrocƚaw Centre for Networking and Supercomputing for providing generous computer time. One of the authors (S.R.) acknowledges the financial support of NCN grant no. UMO-2013/09/B/ST4/00097. Additionally, the project was supported in part by the Wrocƚaw Centre of Biotechnology programme. The Leading National Research Center (KNOW) for years 2014–2018, and a grant of a statutory activity subsidy from Polish Ministry of Science and Technology of Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper is in honor of professor Zdzisƚaw Latajka on his 45 years of research.
This paper belongs to the Topical Collection Zdzislaw Latajka 70th Birthday Festschrift
Electronic supplementary material
Supporting information is available free of charge. NMR spectra of daidzein and puerarin dissolved in methanol-d. Calculated electron spin density for puerarin radicals and calculated transition state for daidzein HAT reaction with OOH• reference.
ESM 1
(DOCX 787 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yi, Y., Adrjan, B., Li, J. et al. NMR studies of daidzein and puerarin: active anti-oxidants in traditional Chinese medicine. J Mol Model 25, 202 (2019). https://doi.org/10.1007/s00894-019-4090-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4090-8