
Abstract
The competition between [2 + 1] and [4 + 1] channels regarding reactions of conjugated nitroalkenes with dichlorocarbene was explored based on B3LYP/6-31G(d) calculations. It was found that, in the case of cycloadditions involving parent nitroethene and its 1-substituted analogs, the [2 + 1] scheme should be treated as possible only from the kinetic process point of view. On the other hand, in similar reactions involving 2-substituted nitroethenes, both channels considered may compete. Additionally, mechanistic aspects of all cycloadditions were analyzed. It was found that the considered [2 + 1]-cycloadditions proceed via a non-polar mechanism with a biradicaloidal transition state (TS), whereas [4 + 1]-cycloadditions proceed via a polar mechanism with a zwitterionic TS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nitro-functionalized carbo- and heterocycles play an important role in modern organic chemistry. This is a consequence of a wide range of further functionalization that may be realized via transformation of nitrocompounds into nitrile N-oxides [1, 2], oximes [3, 4], hydroxylamines [5, 6], nitronates [2, 4], carbonyl compounds [4, 7], aminoalcohols [8, 9] and others [4, 10]; this makes the effective synthesis of many valuable compounds possible.
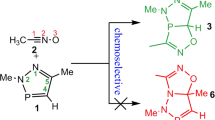
The most universal methodology for preparing nitro-functionalized cyclic compounds is cycloaddition reactions involving respective conjugated nitroalkenes (CNA) [4, 11]. In this way, several four- [12, 13], five -[14,15,16,17], and six-membered [18,19,20,21] nitro-cycles have been prepared. This is possible via [2 + 2], [3 + 2], and [4 + 2] cycloadditions, respectively. All these processes have been realized under relatively mild conditions and with full atomic economy. A detailed literature survey shows, unfortunately, [11], that any examples of [2 + 1]-cycloaddition involving conjugated nitroalkenes (which gave the possibility of the synthesis of nitro-functionalized cyclopropanes) are not known. The work presented here initiates a comprehensive study in this area. In particular, we decided to shed light on the molecular mechanism of model transformations involving a representative series of conjugated nitroalkenes (1a-e) and dichlorocarbene (2) (Scheme 1). Carbene 2 is well known and may be easily generated in the reaction environment [22,23,24,25]. On the other hand, five nitroalkenes (1a-e) characterized by a different type of substitution were selected. All these nitrocompounds may be prepared with good yields [26], can exist as relatively stable compounds, and have been recently tested as components of other type cycloadditions (nitroethene 1a [14, 16, 27], 2-niroprop-1-ene 1b [28], 1-chloro-1-nitroethene 1c [14, 18, 29], E-1-nitroprop-1-ene 1d [28, 30, 31], E-2-chloro-1-nitroethene [31]). We hope that this study will help to further understand the nature of the title reactions and will be useful in organic synthesis.

Structures of selected cycloaddition components
Computational methods
All quantum-chemical calculations were performed using ‘Prometheus’ infrastructure shared by the ACK ‘Cyfronet’ in Kracow, Poland. The B3LYP method is formed through a combination of Becke’s three-parameter hybrid functional [32] and the LYP semi-local correlation function [33] with 6-31G(d) basis set, and was implemented in the Gaussian 09 [34] program. We recently applied an identical level of theory successfully to resolving several different aspects of cycloaddition reactions involving CNAs, such as regioselectivity [14, 17, 21, 30], stereoselectivity [21, 31], molecular mechanism [14, 16, 17, 21, 31, 35], solvent effects [21], substituent effects [14, 21, 35], etc. So, it may be assumed that this approach is adequate for analysis of the issues described above. Subsequently, we also examined higher levels of theory for resolving of the title problem. In these calculations, different types of functionals and basis sets were applied. It was found that all these approaches suggest the same kinetic preferences of reaction channels and similar nature of asynchronicity of transition states (TS). This confirms that the B3LYP/6-31G(d) theory level is adequate for our needs.
All critical structures were optimized using the Berny algorithm [36] and were characterized by frequency calculations. It was found that all addends, molecular complexes (MC), and products (P) had positive Hessian matrices, whereas all TS had one negative eigenvalue in their Hessian matrices. For all TS, intrinsic reaction coordinate (IRC) calculations were performed. The solvent effect was implemented using the polarizable continuum model (PCM) [37]. Global electron density transfer between substructures of the transition state (GEDT) [38] was calculated according to the equation:
where qA is the net charge, and the sum is taken over all the atoms of nitroalkene.
Results of quantum chemical calculations are collected in Tables 1, 2, 3, 4, and 5.
Results and discussion
The [2 + 1]-cycloadition (21CA) process involving CNAs 1a–e and dichlorocarbene 2 (Scheme 1) formally should be resulted in nitrofunctionalized, cyclopropane derivatives (3a–e respectively). A few, recent theoretical works [39,40,41] suggest a one-step mechanism for some [2 + 1]-cycloadditions involving dichlorocarbene. On the other hand, earlier reports [42] suggest a stepwise mechanism, with zwitterionic intermediate. It should be additionally underlined that stepwise mechanisms have been recently documented based on comprehensive experimental and quantum-chemical studies regarding a group of different type cycloadditions involving conjugated nitroalkenes [35, 43, 44]. Therefore, neither one-step nor stepwise mechanisms can be excluded a priori for the reactions studied (Scheme 2).

Possible mechanism of [2 + 1] cycloadditions involving 1 and 2a–e
Next, recent reports confirm that, in some cases, CNAs can be accessed in the addition reactions as hetero-analogs of the 1,3-diene under non-catalytic conditions [45,46,47]. Therefore, the [4 + 1]-cycloaddition (41CA) scheme should also be considered in the analyzed processes (Scheme 3). Finally, we considered six theoretically possible reaction channels (A–E) for reactions 1a-e+2 (Schemes 2 and 3).

Possible mechanism of [4 + 1] cycloadditions involving 1 and 2a–e
Firstly, we decided to analyze the nature of intermolecular interactions during title reactions in the framework of molecular electron density theory (MEDT) [48]. For this purpose, global electrophilicity (ω) for all considered addents were calculated using equations recommended by Parr [49] and Domingo [50, 51]:
As is evident in the light of the data presented in Table 1, in the Domingo scale [50], all considered CNAs should be treated as strongly electrophilic agents. Dichlorocarbene 2 should be localized in the same group. However, its global electrophilicity ω is higher. In particular, the difference between ω values in the reactions involving nitroethene 1a and their alkyl-substituted analogs is equally more than 1.3 eV. Next, for the similar processes involving chloro-substituted CNAs (1c,e), the analogous different is equal about 1 eV. So, in the case of all considered reactions, the polar mechanism can be treatment as allowed.
In the next step, we decided to fully explore of reaction channels leading finally to competitive cycloadducts 3 and 4. It was found that the energetic profile of model 21CA 1a + 2 → 3a suggests a one-step mechanism of addents transformation in toluene solution (Fig. 1, Table 1). In particular, between valleys of individual reagents and product, only one critical point was detected, which is associated with the existence of TS. Reaching this point in the reaction system required rather a small amount of enthalpy (ΔH = 1.5 kcal mol−1). Subsequently, due to the reduction of the entropy of the reaction system, the Gibbs free energy of the activation is higher than ΔH and is equal to 12.8 kcal mol−1. Generally, it is a rather low value, which allowed the reaction to proceed at room temperature.

Reaction profiles for 21CA and 41CA of nitroethene (1a) with dichlorocarbene 2 in toluene solution in the light of B3LYP/6-31G(d) calculations
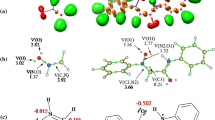
Within the localized TS, two new sigma-bonds are formed: C3–C1 and C2–C1 (Fig. 2, Table 2). The key distances between reaction centers within the TS are, however, distinctly different. In particular, the bond formed with the participation of β carbon atom derived from nitrovinyl moiety (2.271 Å) is more advanced. The second new bond (C3–C1) is more than 0.5 Å longer. This suggests that the considered TS may exhibit a polar, zwitterionic nature. However, this hypothesis can be excluded due to the value of GEDT, which is nearly equal to zero. Therefore, the analyzed TS probably has a biradicaloidal character. The IRC calculations connect the energetic maximum of the transition state, directly with the valleys of individual addents and expected product 3a. Due to Jaque and Murray observation [52, 53], the shape of IRC profiles can be connected with the degree of TS asynchronicity. The formation of 3a is full exothermic, which excludes the reversible nature of considered transformation. It should be mentioned that all attempts to optimize the critical structures that can be associated with hypothetical mechanisms B and C were unsuccessful. Thus, transformation 1a + 2 → 3a is realized via a non-polar one-step mechanism with a highly asynchronous, biradicaloidal TS.

View of key structures for 21CA of nitroethene (1a) with dichlorocarbene 2 in toluene solution in the light of B3LYP/6-31G(d) calculations
Subsequently, in the similar way, we analyzed the competitive reaction channel of the transformation 1a + 2 → 4a. It was found that four critical points exist on the energetic profile of model 41CA (1a + 2 → 4a). These points can be associated with: individual reagents, pre-reaction MC, TS and reaction product 4a (Fig. 1, Table 2). The interactions between starting molecules lead, in the first step, to the formation of the pre-reaction complex MC. This does not require an activation barrier, and is associated with a reduction in enthalpy of about 1 kcal mol−1. It should be underlined that, due to the entropy factor, the ΔG value of MC formation is greater than zero. Therefore, MC cannot exist at room temperature as a stable intermediate. Within the MC, the distances between reactions centers are evidently longer than the typical C–C bonds formed in the TS (Fig. 3). Additionally, MC does not exhibit a CT-complex nature. This was confirmed by the GEDT value (0.00e).

View of key structures for 41CA of nitroethene (1a) with dichlorocarbene 2 in toluene solution in the light of B3LYP/6-31G(d) calculations
Further transformation of the reaction system along the reaction coordinate lead to an energetic maximum. This maximum is associated with the existence of the TS. The transition into TS required an increase in enthalpy of about 6.5 kcal mol−1. In the parallel, due to the reduction of the entropy of the reaction system, the Gibbs free energy of the activation is higher than ΔH, and is equal to >18 kcal mol−1. Within localized TS, two new sigma-bonds are formed: O5–C1 and C2–C1 (Fig. 3, Table 2).
Interestingly, the asynchronicity of this TS is clearly higher than in the case of the TS of transformation 1a + 2 → 3a. In particular, the interatomic distance O5–C1 is 1.8 Å, whereas the C2–C1 distance is 0.85 Å longer. The high asynchronicity is accompanied by transfer between TS substructures. This effect is illustrated by GEDT value, which is 0.15e. So, in contrast to the TS of transformation 1a + 2 → 3a, the considered TS should be treated as a polar, zwitterionic structure. IRC calculations connect the energetic maximum of the TS directly with the valleys of individual addents and expected product 4a. All attempts to optimize critical structures associated with the hypothetical mechanisms E and F were unsuccessful. Thus, the transformation 1a + 2 → 4a was realized via a polar one-step mechanism with a highly asynchronous, zwitterionic TS. Finally, it can be noted that, in the conditions of competition between 21CA and 41CA schemes, the second direct addent transformation should be treated as forbidden from a kinetic point of view (ΔΔG > 5kcal mol−1).
The introduction of a more polar solvent (nitromethane) to the reaction environment does not change the nature of energetic profiles of competitive reaction paths. The quantitative description of critical points is, however, quite different. In particular, the activation barriers are slightly higher; this, however, did not change the kinetic preferences of possible channels of addents transformation. Within the TS of 1a + 2 → 3a transformation, the advancement of both new sigma bonds increased similarly. In consequence, the asynchronicity of TS is rather similar as in the case of similar reaction in toluene environment; this is understandable for non-polar reactions and confirms the nature of 1a + 2 → 3a transformation proposed above.
On the other hand, within the TS of 1a + 2 → 4a transformation, the sigma bond O5–C1 is formed faster than in the case of reaction in toluene, whereas the sigma bond C2–C1 is formed more slowly than in the case of the reaction in toluene. As a consequence, the asynchronicity of TS is higher than in the case of the analogous reaction in a toluene environment, but not enough to enforce a stepwise mechanism. The observed influence of solvent polarity on the synchronicity of the TS structure, however, confirms the polar nature of the 1a + 2 → 4a transformation.
Analogously, the competitive 21CA and 41CA processes involving substituted nitroethenes 1b-e were also explored. It was found that, in all considered cases, the molecular mechanism of addent transformation was very similar to the case of the 1a + 2 reaction. Interestingly, the kinetic preference of possible cycloaddition channels is notably changed to some degree. In particular, reactions involving 2-substituted nitroethenes 2d,e, the 41CA scheme may really compete with the 21CA process. As a consequence, within these reactions, two different cycloaddition products (respective [2 + 1] and [4 + 1] cycloadducts) may form, with some preference for the [2 + 1] pathway. So, the process analyzed may be considered as an exciting alternative to known, generally complicated, protocols [54,55,56] for synthesis of five-membered internal nitronates.
Finally, we also explored in similar way some other reaction systems, including different type substituents at the C1 or C2 position of nitroalkene (Table 5). It was found that the molecular mechanism of all of the explored reactions is very similar. So, the proposed reaction scheme can be assumed as the general mechanism for the vast range of cycloaddition reactions between conjugated nitroalkenes and dichlorocarbene.
Conclusions
DFT investigations at the B3LYP/6-31G(d) level show that molecular mechanisms of competitive [2 + 1] and [4 + 1] cycloadditions of conjugated nitroalkenes with dichlorocarbene are substantially different. In particular, all considered [2 + 1]-cycloadditions proceed via a non-polar mechanism with a biradicaloidal TS, whereas [4 + 1]-cycloadditions proceed via a polar mechanism witha zwitterionic TS. Subsequently, we established that the competition between mentioned cycloaddition schemes is possible only in the case of reactions involving 2-substituted nitroethene analogs. In the case of reactions involving a parent nitroethene as well as its 1-substituted analogs, the [4 + 1] cycloaddition scheme should be treated as forbidden from a kinetic point of view. From a practical point of view, our study suggests that the analyzed processes can be realized under mild conditions, and are a convenient way to prepare nitro-substituted cyclopropane derivatives and (in some cases) five-membered nitronates, which are difficult to realize using other preparation methods.
References
Ajay Kumar K et al (2013) Nitrile oxides: a key intermediate in organic synthesis. IJPCBS 3:91–101
Belenkii L (2007) Nitrile oxides. In: Feuer H (ed) Nitrile oxides, nitrone and nitronates in orgnanic synthesis. Wiley, Hoboken, pp 1–128
Hanson J, Premuzic E (1967) Applications of chromous chloride—II: the reduction of some steroidal nitro-compounds. Tetrahedron 23(10):4105–4110
Ono N (2003) The nitro group in organic synthesis, vol 9. Wiley, New York
Kende AS, Mendoza JS (1991) Controlled reduction of nitroalkanes to alkyl hydroxylamines or amines by samarium diiodide. Tetrahedron Lett 32(14):1699–1702
Rahaim RJ, Maleczka RE (2005) Pd-catalyzed silicon hydride reductions of aromatic and aliphatic nitro groups. Org Lett 7(22):5087–5090
Selvi T, Srinivasan K (2014) Boron trifluoride mediated ring-opening reactions of trans-2-Aryl-3-nitro-cyclopropane-1, 1-dicarboxylates. Synthesis of aroylmethylidene malonates as potential building blocks for heterocycles. J Org Chem 79(8):3653–3658
Bergmeier SC (2000) The synthesis of vicinal amino alcohols. Tetrahedron 56(17):2561–2576
Soengas RG, Silva AM (2012) Indium-catalyzed Henry-type reaction of aldehydes with bromonitroalkanes. Synlett 23(6):873–876
Paul B et al (2017) Tandem transformation of nitro compounds into N-methylated amines: greener strategy for the utilization of methanol as a methylating agent. ChemSusChem 10(11):2370–2374
Łapczuk-Krygier A, Kącka-Zych A, Kula K (2019) Recent progress in the field of cycloaddition reactions involving conjugated nitroalkenes. Current Chem Lett 8(1):13–38
Mohr L-M, Bach T (2017) Intermolecular [2+ 2] photocycloaddition of β-Nitrostyrenes to olefins upon irradiation with visible light. Synlett 28(20):2946–2950
Albrecht Ł et al (2012) Asymmetric organocatalytic formal [2+ 2]-cycloadditions via bifunctional H-bond directing dienamine catalysis. J Am Chem Soc 134(5):2543–2546
Jasiński R, Jasińska E, Dresler E (2017) A DFT computational study of the molecular mechanism of [3+ 2] cycloaddition reactions between nitroethene and benzonitrile N-oxides. J Mol Model 23(1):13
Mirosław B et al (2018) Regiospecific formation of the nitromethyl-substituted 3-phenyl-4, 5-dihydroisoxazole via [3+ 2] cycloaddition. Monatsh Chem 149(10):1877–1884
Jasiński R (2015) In the searching for zwitterionic intermediates on reaction paths of [3+ 2] cycloaddition reactions between 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide and polymerizable olefins. RSC Adv 5(122):101045–101048
Jasiński R, Mróz K, Kącka A (2016) Experimental and theoretical DFT study on synthesis of sterically crowded 2,3,3,4) 5-Tetrasubstituted-4-nitroisoxazolidines via 1, 3-dipolar cycloaddition reactions between Ketonitrones and conjugated Nitroalkenes. J Heterocyclic Chem 53(5):1424–1429
Jasiński R (2017) One-step versus two-step mechanism of Diels-Alder reaction of 1-chloro-1-nitroethene with cyclopentadiene and furan. J Mol Graph Model 75:55–61
Łapczuk-Krygier A, Jasiński R (2014) The crystal structure of (1RS, 4RS, 5RS, 6SR)-5-cyano-5-nitro-6-phenyl-bicyclo [2.2.1] hept-2-ene. Crystallogr Rep 59(7):961–963
Jasiński R (2016) First example of stepwise, zwitterionic mechanism for bicyclo [2.2.1] hept-5-ene (norbornene) formation process catalyzed by the 1-butyl-3-methylimidazolium cations. Monatsh Chem 147(7):1207–1213
Jasiński R (2018) β-Trifluoromethylated nitroethenes in Diels-Alder reaction with cyclopentadiene: a DFT computational study. J Fluor Chem 206:1–7
Khlebnikov A et al (1990) 1, 3-dipolar cycloaddition reactions of ylides formed from pyridines and dichlorocarbene. Chem Heterocycl Compd 26(3):304–311
Rajendran V, Wang M-L (2008) Dichlorocarbene addition to allyl phenyl ether under phase-transfer catalysis conditions—a kinetic study. J Mol Catal A Chem 288(1–2):23–27
Egorova AY, Sedavkina VA, Timofeyeva ZY (2000) Interaction of 1, 5-substituted pyrrolin-2-ones with dichlorocarbene under phase transfer catalysis conditions. Molecules 5(10):1082–1084
Mohamadi F, Still WC (1986) Dichlorocarbene cyclopropanation of allylic alcohols. Tetrahedron Lett 27(8):893–896
Novikov S, et al (1974) Khimiya alifaticheskikh i alitsiklicheskikh nitrosoedinenii [Chemistry of Aliphatic and Alicyclic Nitro Compounds]. Khimiya, Moscow, pp 239
Kula K, Łapczuk-Krygier A (2018) A DFT computational study on the [3+ 2] cycloaddition between parent thionitrone and nitroethene. Current Chem Lett 7(1):27–34
Jasiński R (2015) Synthesis and properties of azoles and their derivatives. Part 70. Nitroallylic systems in [2+ 3] cycloaddition reactions with nitrones: a DFT computational study. J Heterocyclic Chem 52(1):185–192
Jasiński R (2015) A stepwise, zwitterionic mechanism for the 1, 3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalysed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett 56(3):532–535
Jasiński R et al (2014) Regio-and stereoselectivity of polar [2+ 3] cycloaddition reactions between (Z)-C-(3, 4, 5-trimethoxyphenyl)-N-methylnitrone and selected (E)-2-substituted nitroethenes. Open Chem 12(5):586–593
Jasiński R (2012) Exploration of regiospecificity phenomenon in [2+ 3] cycloaddition reactions between arylnitrones and trans-substituted nitroethenes on the basis of the reactivity indices theory. Curr Chem Lett 1(4):157–162
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7):5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785
Frisch M et al (2009) Gaussian 09 software. Gaussian Inc, Wallingford
Jasiński R et al (2017) A full regioselective and stereoselective synthesis of 4-Nitroisoxazolidines via stepwise [3+ 2] cycloaddition reactions between (Z)-C-(9-Anthryl)-N-arylnitrones and (E)-3, 3, 3-Trichloro-1-nitroprop-1-ene: comprehensive experimental and theoretical study. J Heterocyclic Chem 54(6):3314–3320
Peng C et al (1996) Using redundant internal coordinates to optimize equilibrium geometries and transition states. J Comput Chem 17(1):49–56
Cossi M et al (2003) Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comput Chem 24(6):669–681
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4(61):32415–32428
Petrushenko IK (2014) [2+ 1] cycloaddition of dichlorocarbene to finite-size graphene sheets: DFT study. Monatsh Chem 145(6):891–896
Zeroual A, Benharref A, El Hajbi A (2015) Theoretical study of stereoselectivity of the [1+ 2] cycloaddition reaction between (1S, 3R, 8S)-2, 2-dichloro-3, 7, 7, 10-tetramethyltricyclo [6, 4, 0, 0 1.3] dodec-9-ene and dibromocarbene using density functional theory (DFT) B3LYP/6-31G*(d). J Mol Model 21(3):44
Lu X, Tian F, Zhang Q (2003) The [2+ 1] cycloadditions of dichlorocarbene, silylene, germylene, and oxycarbonylnitrene onto the sidewall of armchair (5, 5) single-wall carbon nanotube. J Phys Chem B 107(33):8388–8391
Von E, Doering W, Henderson Jr WA (1958) The electron-seeking demands of dichlorocarbene in its addition to olefins. J Am Chem Soc 80(19):5274–5277
Jasiński R et al (2017) Unexpected course of reaction between (E)-2-aryl-1-cyano-1-nitroethenes and diazafluorene: why is there no 1, 3-dipolar cycloaddition? Monatsh Chem 148(5):909–915
Jasiński R (2018) Competition between one-step and two-step mechanism in polar [3+ 2] cycloadditions of (Z)-C-(3, 4, 5-trimethoxyphenyl)-N-methyl-nitrone with (Z)-2-EWG-1-bromo-1-nitroethenes. Comput Theor Chem 1125:77–85
Jasiński R et al (2014) An experimental and theoretical study of the hetero Diels–Alder reactions between (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether: one-step or zwitterionic, two-step mechanism? React Kinet Mech Catal 113(2):333–345
Tohda Y et al (1988) Synthesis and a novel fragmentation of 6-Alkoxy-5, 6-dihydro-4 H-1, 2-oxazine 2-oxide. Bull Chem Soc Jpn 61(2):461–465
Jasiński R (2014) Searching for zwitterionic intermediates in hetero diels–alder reactions between methyl α, p-dinitrocinnamate and vinyl-alkyl ethers. Comput Theor Chem 1046:93–98
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21(6):748
Parr RG, Szentpaly Lv, Liu S (1999) Electrophilicity index. J Am Chem Soc 121(9):1922–1924
Pérez P et al (2003) Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1, 3-dipolar cycloaddition reactions. Tetrahedron 59(17):3117–3125
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J Org Chem 73(12):4615–4624
Yepes D et al (2014) Complementarity of reaction force and electron localization function analyses of asynchronicity in bond formation in Diels–Alder reactions. Phys Chem Chem Phys 16(14):6726–6734
Murray JS et al (2015) Insights into some Diels–Alder cycloadditions via the electrostatic potential and the reaction force constant. Comput Theor Chem 1053:270–280
Ioffe S et al (1973) Silylation of nitro-compounds. 3. Dinitrocarbene formation in trimethylsilylation of trinitromethane derivatives. Zh Org Khim 9(5):905–912
Kunetsky RA et al (2003) New approach for the synthesis of isoxazoline-N-oxides. Org Lett 5(25):4907–4909
Kunetsky RA et al (2006) General method for the synthesis of isoxazoline N-oxides from aliphatic nitro compounds. Synthesis 2006(13):2265–2270
Acknowledgment
The regional computer center “Cyfronet” in Krakow (‘Prometheus’ cluster, project ‘Heterocycles’) is thanked for the allocation of computing time.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Alnajjar, R.A., Jasiński, R. Competition between [2 + 1]- and [4 + 1]-cycloaddition mechanisms in reactions of conjugated nitroalkenes with dichlorocarbene in the light of a DFT computational study. J Mol Model 25, 157 (2019). https://doi.org/10.1007/s00894-019-4006-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4006-7