
Abstract
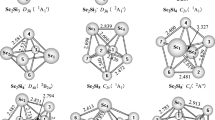
In this work, the low-lying states of several isomers of ScSi n −/0 (n = 4–6) were investigated with the B3LYP functional and CASPT2 method. The ground states of the anionic clusters were predicted to be the singlet states of the trigonal bipyramid ScSi4 − (A-ScSi4 −), the face-capped trigonal bipyramid ScSi5 − (A-ScSi5 −), and the pentagonal bipyramid ScSi6 − (A-ScSi6 −) isomer. Based on the anionic ground states, all the relevant electron detachment processes were identified. The corresponding adiabatic and vertical detachment energies (ADEs and VDEs) of the anionic clusters were computed at the CASPT2 level. The calculated results were used to interpret all the important features in the photoelectron spectra of ScSi n − (n = 4–6) clusters. Franck-Condon factor simulations were also performed based on the B3LYP geometries, vibrational frequencies, and normal modes to explain the shapes of the first bands in the spectra.









Similar content being viewed by others

References
Koyasu K, Akutsu M, Mitsui M, Nakajima A (2005) Selective formation of MSi16 (M = Sc, Ti, and V). J Am Chem Soc 127(14):4998–4999. https://doi.org/10.1021/ja045380t
Koyasu K, Atobe J, Akutsu M, Mitsui M, Nakajima A (2007) Electronic and geometric stabilities of clusters with transition metal encapsulated by silicon. J Phys Chem A 111 (1):42–49. doi:https://doi.org/10.1021/jp066757f
Koyasu K, Atobe J, Furuse S, Nakajima A (2008) Anion photoelectron spectroscopy of transition metal- and lanthanide metal-silicon clusters: MSi n − (n = 6-20). J Chem Phys 129(21):214301. https://doi.org/10.1063/1.3023080
Xu H-G, Wu M-M, Zhang Z-G, Sun Q, Zheng W-J (2011) Structural and bonding properties of ScSi n − (n = 2 ~ 6) clusters: photoelectron spectroscopy and density functional calculations. Chin Phys B 20(4):043102
Xu H-G, Zhang Z-G, Feng Y, Zheng W (2010) Photoelectron spectroscopy and density-functional study of Sc2Si n − (n = 2-6) clusters. Chem Phys Lett 498(1–3):22–26. https://doi.org/10.1016/j.cplett.2010.08.027
DeYonker NJ, Williams TG, Imel AE, Cundari TR, Wilson AK (2009) Accurate thermochemistry for transition metal complexes from first-principles calculations. J Chem Phys 131(2):024106. https://doi.org/10.1063/1.3160667
Mayhall NJ, Raghavachari K, Redfern PC, Curtiss LA (2009) Investigation of Gaussian4 theory for transition metal thermochemistry. J Phys Chem A 113(17):5170–5175. https://doi.org/10.1021/jp809179q
Xiao C, Abraham A, Quinn R, Hagelberg F, Lester WA (2002) Comparative study on the interaction of scandium and copper atoms with small silicon clusters. J Phys Chem A 106(46):11380–11393. https://doi.org/10.1021/jp021668y
Lu J, Yang J, Kang Y, Ning H (2014) Probing the electronic structures and properties of neutral and anionic ScSi n (0,−1) (n = 1-6) clusters using ccCA-TM and G4 theory. J Mol Model 20(2):1–12. https://doi.org/10.1007/s00894-014-2114-y
Borshch N, Kurganskii S (2014) Geometric structure, electron-energy Spectrum, and growth of anionic scandium-silicon clusters ScSi n − (n = 6 - 20). J Appl Phys 116(12):124302. https://doi.org/10.1063/1.4896528
He J, Wu K, Liu C, Sa R (2009) Stabilities of 3d transition-metal doped Si14 clusters. Chem Phys Lett 483(1–3):30–34. https://doi.org/10.1016/j.cplett.2009.10.052
Ulises Reveles J, Khanna SN (2006) Electronic counting rules for the stability of metal-silicon clusters. Phys Rev B 74(3):035435
Torres MB, Balbás LC (2007) Relative stability of Si n and Si n Sc− clusters in the range n = 14 - 18. Eur Phys J D 43(1):217–220. https://doi.org/10.1140/epjd/e2007-00086-8
Tran QT, Tran VT (2016) Quantum chemical study of the geometrical and electronic structures of ScSi3 −/0 clusters and assignment of the anion photoelectron spectra. J Chem Phys 144(21):214305. https://doi.org/10.1063/1.4953082
Pham LN, Nguyen MT (2016) Electronic structure of neutral and anionic scandium Disilicon ScSi2 −/0 clusters and the related anion photoelectron Spectrum. J Phys Chem A 120(47):9401–9410. https://doi.org/10.1021/acs.jpca.6b09067
Tran VT, Tran QT (2016) Geometrical and electronic structures of MnS3 −/0 clusters from computational chemistry and photoelectron spectroscopy. J Phys Chem A 120(20):3670–3676. https://doi.org/10.1021/acs.jpca.6b02631
Tran VT, Tran QT (2016) Quantum chemical study of the low-lying electronic states of VSi3 −/0 clusters and interpretation of the anion photoelectron spectrum. J Phys Chem A 120(29):5950–5957. https://doi.org/10.1021/acs.jpca.6b05653
Tran VT, Iftner C, Hendrickx MFA (2013) Quantum chemical study of the electronic structures of MnC2 −/0 clusters and interpretation of the anion photoelectron spectra. Chem Phys Lett 575 (0):46–53. doi:https://doi.org/10.1016/j.cplett.2013.04.079
Tran VT, Hendrickx MFA (2014) Molecular and electronic structures of the NbC2 −/0 clusters through the assignment of the anion photoelectron spectra by quantum chemical calculations. Chem Phys Lett 609:98–103. doi:https://doi.org/10.1016/j.cplett.2014.06.046
Tran VT, Hendrickx MFA (2011) Assignment of the photoelectron spectra of FeS3 −/0 by density functional theory, CASPT2, and RCCSD(T) calculations. J Phys Chem A 115(47):13956–13964. https://doi.org/10.1021/jp208824b
Tran VT, Hendrickx MFA (2013) Molecular structures for FeS4 −/0 as determined from an ab initio study of the anion photoelectron spectra. J Phys Chem A 117(15):3227–3234. https://doi.org/10.1021/jp401343j
Borrelli R, Peluso A (2003) Dynamics of Radiationless transitions in large molecular systems: a Franck-Condon-based method accounting for displacements and rotations of all the normal coordinates. J Chem Phys 119(16):8437–8448. https://doi.org/10.1063/1.1609979
Borrelli R, Capobianco A, Peluso A (2013) Franck-Condon factors—computational approaches and recent developments. Can J Chem 91(7):495–504. https://doi.org/10.1139/cjc-2012-0518
Valiev M, Bylaska EJ, Govind N, Kowalski K, Straatsma TP, Van Dam HJJ, Wang D, Nieplocha J, Apra E, Windus TL, de Jong WA (2010) NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput Phys Commun 181(9):1477–1489. https://doi.org/10.1016/j.cpc.2010.04.018
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297–3305
Aquilante F, Autschbach J, Carlson RK, Chibotaru LF, Delcey MG, De Vico L, Fdez, Galván I, Ferré N, Frutos LM, Gagliardi L, Garavelli M, Giussani A, Hoyer CE, Li Manni G, Lischka H, Ma D, Malmqvist PÅ, Müller T, Nenov A, Olivucci M, Pedersen TB, Peng D, Plasser F, Pritchard B, Reiher M, Rivalta I, Schapiro I, Segarra-Martí J, Stenrup M, Truhlar DG, Ungur L, Valentini A, Vancoillie S, Veryazov V, Vysotskiy VP, Weingart O, Zapata F, Lindh R (2016) Molcas 8: new capabilities for multiconfigurational quantum chemical calculations across the periodic table. J Comput Chem 37(5):506–541. https://doi.org/10.1002/jcc.24221
Balabanov NB, Peterson KA (2005) Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc-Zn. J Chem Phys 123(6):064107. https://doi.org/10.1063/1.1998907
Woon DE, Dunning TH (1993) Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys 98(2):1358–1371. https://doi.org/10.1063/1.464303
Ishikawa Y, Vilkas MJ (2001) Relativistic quantum mechanics of many-electron systems. J Mol Struc THEOCHEM 573(1–3):139–169. https://doi.org/10.1016/S0166-1280(01)00540-1
Reiher M, Wolf A (2004) Exact decoupling of the Dirac Hamiltonian. I. General theory. J Chem Phys 121(5):2037–2047. https://doi.org/10.1063/1.1768160
Reiher M, Wolf A (2004) Exact decoupling of the Dirac Hamiltonian. II. The Generalized Douglas-Kroll-Hess Transformation up to Arbitrary Order. J Chem Phys 121(22):10945–10956. https://doi.org/10.1063/1.1818681
Aquilante F, Lindh R, Bondo Pedersen T (2007) Unbiased auxiliary basis sets for accurate two-electron integral approximations. J Chem Phys 127(11):114107. https://doi.org/10.1063/1.2777146
Aquilante F, Malmqvist P-Å, Pedersen TB, Ghosh A, Roos BO (2008) Cholesky decomposition-based multiconfiguration second-order perturbation theory (CD-CASPT2): application to the spin-state energetics of CoIII(diiminato)(NPh). J Chem Theory Comput 4(5):694–702. https://doi.org/10.1021/ct700263h
Aquilante F, Pedersen TB, Lindh R, Roos BO, Sánchez de Merás A, Koch H (2008) Accurate ab initio density fitting for multiconfigurational self-consistent field methods. J Chem Phys 129(2):024113. https://doi.org/10.1063/1.2953696
Forsberg N, Malmqvist P-Å (1997) Multiconfiguration perturbation theory with imaginary level shift. Chem Phys Lett 274(1–3):196–204. https://doi.org/10.1016/S0009-2614(97)00669-6
Tran VT, Hendrickx MFA (2011) A CASPT2 description of the electronic structures of FeO3 −/0 in relevance to the anion photoelectron spectrum. J Chem Theory Comput 7(2):310–319. https://doi.org/10.1021/Ct1005246
Acknowledgment
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.06-2016.16.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nguyen, M.T., Tran, Q.T. & Tran, V.T. A CASSCF/CASPT2 investigation on electron detachments from ScSi n − (n = 4–6) clusters. J Mol Model 23, 282 (2017). https://doi.org/10.1007/s00894-017-3461-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3461-2