
Abstract
The internal rotation in ammonia borane (AB) was studied on the basis of natural orbitals for chemical valence (NOCV) and eigenvectors for Pauli repulsion (NOPR). We found that the total hyperconjugation stabilization (ca. 5 kcal mol−1), based on the charge transfer from the occupied σ (B–H) orbitals into the empty σ*(N–H), slightly favors the staggered conformation over the eclipsed one; however, the barrier to internal rotation in ammonia borane can be understood predominantly in a ‘classical’ way, as originating from the steric (Pauli) repulsion contributions (of the kinetic origin) that act solely between N–H and B–H bonds. Repulsion between the lone pair of ammonia and the adjacent B–H bonds was found to be dominant in absolute terms; however, it does not determine the rotational barrier. Similar conclusions on the role of CH↔HC repulsion appeared to be valid for isoelectronic ethane.

Pauli (kinetic) repulsion acting between the N-H and B-H bonds of ammonia borane
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the most important goals of theoretical chemistry is to understand the origin of conformational changes in molecules [1, 2]. In order to achieve this goal many methods can be applied to the description of electronic structures: molecular orbitals (MOs) [3, 4], localized molecular orbitals (LMOs) [5–7], bond orders [8–17], atoms in molecules (AIM) [18], Fermi hole [19], kinetic energy and information theory based quantities [20–22], and various charge and energy decomposition schemes [23–28]. A useful and elegant approach suitable for description of energy profiles of chemical reactions was proposed by Torro-Labbe and coworkers [29] based on the reaction force concept. Bickelhaupt and Zeist [30] proposed the “activation strain model”, which also appears to be very useful in the analysis of chemical reactions.
Rotation around a single carbon–carbon bond is one of the most important conformational transitions in organic chemistry [31, 32]. A typical example is ethane, which exhibits staggered and eclipsed conformations; the former minimum energy structure is more stable than the transition state eclipsed structure by ∼3.0 kcal mol−1 [33]. The classical and intuitive explanation of the barrier suggested in organic textbooks is based on the steric repulsion between C–H bonds, which is dominant in the eclipsed structure [34]. An alternative explanation is based on hyperconjugation stabilization, which is stronger in the staggered conformation [35–38]. However, as pointed out by Mulliken [35], hyperconjugation effect should have only a minor influence on the barrier. Bader et al. [39] stated that the barrier to rotation in ethane can be related to the polarization of charge density along the carbon–carbon bond. Goodman and coworkers [40] have shown, based on the natural bond orbitals (NBO) method, that ethane’s staggered conformation is the result of hyperconjugation. This point of view was also presented by Weinhold [41]. Goodman’s results based on the NBO method have been challenged by the work of Bickelhaupt and Baerends [42] based on the model of a chemical bond originating from fragmented molecular orbitals; according to these results the internal rotational barrier in ethane is due to Pauli repulsion acting between the CH bonds of opposite CH3 units. Subsequent calculations by Mo and coworkers [44, 45] and then by Pendas et al. [43], confirmed the classical, steric-based interpretation of the barrier; in addition, the former authors have shown that hyperconjugation stabilizes the staggered conformer only by about 4 kJ mol−1 relative to the eclipsed form [45]. A very elegant recent paper by Mo and Gao [46] provided a compact overview of the most important studies on this subject; the main conclusion is that the internal rotational barrier in ethane is due predominantly to steric effects acting between C–H bonds, with minor participation stemming from hyperconjugation.
We have recently developed the ETS-NOCV scheme [47–50] by combining the extended transition state (ETS) [25, 26] energy decomposition approach with the natural orbitals for chemical valence (NOCV) method [51–57]. ETS-NOCV has proved suitable for qualitative and quantitative description of the crucial components (σ, π, δ, etc.) that constitute various types of chemical bonds [58–61]. In addition, the energy profiles of some chemical reactions can be also characterized [62, 63]. More importantly for this study, it was shown that NOCV representation allows for qualitative and quantitative description of hyperconjugation effects [47, 48]. Furthermore, the ETS energy decomposition scheme provides quantitative information on the Pauli repulsion effects [2, 25, 26].
Therefore, the main goal of this article was to apply for the first time the ETS-NOCV charge and energy decomposition scheme to analysis of the internal rotation in ammonia borane. Hyperconjugation and steric factors will be discussed in a detailed way. It should be noted that ammonia borane is considered nowadays as one of the most promising hydrogen storage materials. In addition, it was already proven that ammonia borane exhibits dissimilar features as compared to isoelectronic ethane [64]. Furthermore, the present study sheds additional qualitative and quantitative light on the steric repulsion in ammonia borane by decomposition of total Pauli repulsion into specific contributions stemming from different symmetry (σ and π). In order to achieve this goal, we defined for the first time the eigenvectors for Pauli repulsion; in this representation, one can thus discuss the Pauli repulsion components originating from different symmetries. For comparison, similar analyses will be performed for ethane.
Computational details
All DFT calculations presented here were based on the Amsterdam Density Functional (ADF 2009.01) program [2, 65–68] in which the ETS-NOCV scheme was implemented [47–63]. The Becke-Perdew exchange-correlation functional [69, 70] was applied (BP86). A standard triple-zeta STO basis containing two sets of polarization functions (TZ2P) was adopted for all atoms. The contours of deformation densities were plotted based on ADF-GUI interface [71].
Computational methods
Our analysis is based on the ETS-NOCV approach, which is a combination of the extended transition state (ETS) [25, 26] method with the natural orbitals for chemical valence (NOCV) scheme [51–57].
The basic concept of the ETS scheme involves partitioning of the total bonding energy ΔE total between interacting fragments into four components:
The first component, ΔE dist, referred to as the distortion term, represents the amount of energy required to promote the separated fragments from their equilibrium geometry to the structure they will take up in the combined molecule; it can also be seen as strain energy. The second term, ΔE elstat, corresponds to the classical electrostatic interaction between the promoted fragments as they are brought to their positions in the final complex. The third term, ΔE Pauli, accounts for the repulsive Pauli interaction between occupied orbitals on the two fragments in the combined molecule. It is calculated as the difference between the energies of orthogonalized and non-orthogonalized fragments [2, 25]. Finally, the last stabilizing term, ΔE orb, represents the interactions between the occupied molecular orbitals of one fragment with the unoccupied molecular orbitals of the other fragment as well as the mixing of occupied and virtual orbitals within the same fragment (inner-fragment polarization). This energy term, ΔE orb, may be linked to the electronic bonding effect coming from the formation of a chemical bond (Eq. 2).
The NOCV are eigenvectors that diagonalize deformation density matrix ∆Porb = Pmolecule − P0, where P0 corresponds to the sum of density matrices for orthogonalized fragments; it has been shown that the natural orbitals for chemical valence pairs (ψ-k,ψk) decompose the deformation density Δρ orb into NOCV-contributions, Δρ korb :
where ν k and M are the NOCV eigenvalues and the number of basis functions, respectively. Visual inspection of deformation density plots (Δρ korb ) helps to attribute symmetry and the direction of the charge flow. In addition, information gained from the analysis of deformation density plots can be enriched by providing the energetic estimations, ΔE korb , for each Δρ korb within ETS-NOCV scheme:
where F TSi,i are diagonal Kohn-Sham matrix elements defined over NOCV with respect to the transition state density (at the midpoint between density of the molecule and the sum of fragment densities). The above components ΔE korb provide the energetic estimation of Δρ korb that may be related to the importance of a particular electron flow channel for the bonding between the considered molecular fragments.
In the present study, in analogy to NOCVs, we defined for the first time the natural orbitals (eigenvectors) for Pauli repulsion, ϕk, that diagonalize the Pauli deformation density matrix, ∆PPauli = P0−Pisolated, where Pisolated is the sum of density matrices for non-orthogonalized fragments, whereas P0 correspond to the sum of density matrices for orthogonalized fragments. Such eigenvectors decompose the total Pauli deformation density, ∆ρPauli=ρ0 (orthogonalized-fragments)−ρ (non-orthogonalized-fragments), into the NOCV-like contributions (Δρ Paulik ) (in analogy to Eq. 1):
The total charge transferred in this channel can be considered as:
The present study characterized not only the total values of Pauli repulsion (ΔE Pauli) in ammonia borane based on the original ETS scheme (Eq. 1) but, in addition, provided a more detailed picture by analyses of both the Pauli repulsion contributions Δρ Paulik (Eq. 3) and the corresponding quantitative charge estimations Δq Paulik (Eq. 4). This approach (Eqs. 3, 4) was implemented by one of us in the home version of ADF2009.01. At present, the energetic Pauli repulsion contributions (ΔE kPauli ) from Δρ Paulik (calculated in an analogous way to Eq. 2) are unavailable. Hence, we focused our attention on the quantitative measures of Δρ Paulik based on Eq. 4. Red areas of deformation density channels correspond to charge depletion, whereas blue indicates charge accumulation upon bond formation.
Due to the fact that the steric interaction, which is a non-observable quantity [72], is very often attributed in the literature to Pauli repulsion quantum effect [2, 43, 46], we use both terms interchangeably throughout the text. Finally, we should note that Pauli repulsion is one of the bonding components in various energy decomposition schemes; hence, we believe that a more detailed description of this term based on Eqs. 3, 4, could be of wide interest. It is very important to point out that the main source of the Pauli repulsion is related to an increase in the kinetic energy contribution; so we could also refer to the Pauli repulsion term as kinetic repulsion due to the Pauli exclusion principle [2]. Such a concept, which relates the steric repulsion to the ‘kinetic energy pressure’ has already been put forward by various authors [73, 74]. In addition, the Pauli repulsion contribution appears to qualitatively correlate very well with the experimental Taft’s steric parameters [75].
Results and discussion
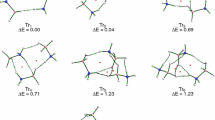
We will start with a brief description of the bonding situation in the most stable staggered conformation (S) of ammonia borane (Fig. 1). It can be seen from Table 1 that the bond dissociation energy (−ΔE total) amounts to 31.94 kcal mol−1 (BP86/TZ2P). This value fits well to the experimental enthalpy estimated by Haaland (31.1 ± 1 kcal mol−1 [76, 77]) as well as to other theoretical estimations [48, 78–81]. In line with previous studies [48, 78, 82–85], we found a slight dominance (by ∼0.7 kcal mol−1) of the electrostatic stabilization over the orbital interaction term (Table 1). Decomposition of the latter stabilizing term into NOCV-based deformation density channels leads to the conclusion that donation (Δρ σorb ) from the lone electron pair of ammonia to the lowest unoccupied orbital of BH3 is by far most dominant (ΔE σorb = − 66.32 kcal mol−1) as compared to the two hyperconjugation contributions, Δρ hyp1orb , Δρ hyp2orb ; the corresponding orbital interaction stabilizations are ΔE hyp1orb = ΔE hyp2orb = − 2.30 kcal mol−1 (Fig. 2). The latter two degenerated contributions stem from charge transfer from the occupied σ (B–H) orbitals into the empty σ*(N–H) (Fig. 2). It is noteworthy that, in the isoelectronic ethane, the sum of stabilization arising from the two orthogonal hyperconjugation components was found to be significantly stronger (∼10 kcal mol−1) [46, 85].

Energy profile for internal rotation in ammonia borane. B–N bond lengths (in Å) are indicated

Dominant natural orbitals for chemical valence (NOCV)-based deformation density channels, Δρ σorb , Δρ hyp1orb , Δρ hyp1orb , with the corresponding orbital interaction energies for the alternative isomers of ammonia borane. The contour value is |∆ρ| = 0.005 a.u. for Δρ σorb , whereas for remaining hyperconjugation contributions 0.001 a.u. was applied. Blue/red contours correspond to accumulation/depletion of electron density. S Fully optimized staggered isomer, E s geom eclipsed structure in the staggered geometry, E fully optimized eclipsed isomer
It is clear from Fig. 1 that rotation from the staggered to the eclipsed form leads to a change in energy, by 1.93 kcal mol−1. This barrier agrees quite well with the experimental value of 2.07 kcal mol−1 determined based on microwave spectra [86], and with other high level computations [87]. It is very important to point out that when going from the staggered (S) to the eclipsed isomer (E), one observes a notable stretch of the B–N bond, by ∼0.03 Å. Such elongation leads expectedly to a significant decrease in Pauli repulsion, by 7.27 kcal mol−1; at the same time the electrostatic (ΔEelstat) and orbital interaction (ΔEorb) contributions become less stabilizing, by 4.14 kcal mol−1 and 4.69 kcal mol−1, respectively (see Table 1 and the blue line in Fig. 3). From the examples of ethane [42] or biphenyl [88], it is known that this type of elongation when going from one isomer to the other is due to the steric (Pauli) repulsion. As indicated in a series of recent works [42–46, 88, 89], in order to estimate and characterize the forces leading to such elongation, one must first consider rigid rotation from the staggered to the eclipsed conformation; we have labeled such eclipsed conformation (in the staggered geometry) as E s geom. We can clearly see now from Table 1 and Fig. 3 (the orange curve), that an increase in the Pauli repulsion contribution, by 2.11 kcal mol−1, is noted when going from S to E s geom; it is important to note that the remaining bonding components are practically unchanged. A similar trend, i.e., the maximum Pauli repulsion in ammonia borane with the dihedral angle ∠(H–B–N–H) = 0.0, is noted when considering the rigid rotation from the geometry of the eclipsed structure to the staggered one (S e geom) (gray curve in Fig. 3). Thus, the Pauli (steric) repulsion contribution is responsible for stretching of the B–N bond and, accordingly, for the rotational barrier in ammonia borane; the analogous situation holds true for the ethane molecule, as demonstrated first by Bickelhaupt et al. [42] and then by others [43–46]. An increased kinetic repulsion (the main source of the Pauli term) in the E s geom geometry is related through the virial theorem to the existence of repulsive forces acting predominantly on nitrogen and boron nuclei [18]. It must be added that hypercongutation stabilizations stemming from the charge transfer from the occupied σ (B–H) orbitals into the empty σ*(N–H) (Δρ hyp1orb , Δρ hyp2orb ), favors the staggered conformation (Fig. 2), although the effect is minor (∼0.4 kcal mol−1) compared to changes in the remaining bonding contributions (Table 1). A quantitatively similar effect is observed for the change in the energy distortion contribution (ΔE dist) (Table 1).

Pauli repulsion energies in the alternative isomers of ammonia borane. Blue Fully relaxed structures; orange staggered geometry; gray eclipsed geometry. S Fully optimized staggered isomer, E s geom eclipsed structure in the staggered geometry, S e geom staggered structure in the eclipsed geometry, E fully optimized eclipsed isomer
Let us now focus our attention on detailed changes in the Pauli repulsion contributions in the three ammonia borane isomers, S, E s geom and E. Figure 4 presents the total Pauli deformation density contours (ΔρPauli) together with the corresponding energy values (ΔE Pauli).

Contours of the total Pauli deformation density together with the corresponding energies. In addition, charge-based estimations are presented based on Eq. 4. The blue/red contours correspond to accumulation/depletion of electron density due to the Pauli exclusion principle
As already stated, the S→ E s geom transition leads to a jump in the Pauli term that it is then ‘relieved’ by elongation of the B–N bond in step E s geom→E. The important question that arises at this point is how the total Pauli repulsion is ‘distributed’ between NH3 and BH3 fragments. An analysis of function ΔρPauli leads to the observation that electrons are removed from the N–B binding region (in fact, it is a manifestation of the Pauli exclusion principle [2, 90]); although one can see that the red lobes extend also to the areas of NH↔HB interaction. However, such contours do not allow us to extract information on whether the total changes in Pauli repulsion are determined by the repulsive interaction between the lone electron pair of ammonia with the occupied σ (B–H) orbitals or directly by ‘classical’ N-H↔H-B repulsion [an interaction between the occupied σ (B–H) orbitals with the occupied σ (N–H)]. In order to obtain such separated information, we have decomposed total Pauli repulsion ΔρPauli into the contributions (Δρ Paulik ) according to Eq. 3. The three leading Pauli deformation density channels, Δρ Pauli1 , Δρ Pauli2 , Δρ Pauli3 , together with the corresponding quantitative charge estimations (Eq. 4) are presented in Fig. 5. It should be noted that the total charge, ΔQ Pauliglobal = Δq Pauli1 + Δq Pauli2 + Δq Pauli3 , that is removed from the H3N–BH3 binding region correlates well with the trend based on the Pauli repulsion energy (Table 1, Fig. 4). Qualitative inspection of the contours Δρ Paulii leads to the important observation that the first channel (Δρ Pauli1 ) corresponds solely to the interaction between the lone electron pair of ammonia with the B–H bonds, whereas the two latter orthogonal contributions (Δρ Pauli2 , Δρ Pauli3 ) show NH↔HB repulsion (Fig. 5). More importantly, quantitative analysis of the charge depletion, based on the eigenvalues (Eq. 3), leads to the conclusion that, when going from S→ E s geom, the major changes (by 0.0148 a.u.) are within the second and third values of Δq Pauli2 , Δq Pauli3 . The repulsion Δρ Pauli1 characterized by Δq Pauli1 remains unchanged. Once going to the relaxed eclipsed structure, the Pauli contribution is further ‘relieved’ (Δρ Paulii values decrease in line with ΔEPauli). These results show that an increase in the total repulsion in the eclipsed conformation compared to staggered (S→ E s geom) is determined solely by the NH↔HB repulsion (of the kinetic origin) due to the Pauli exclusion principle (an interaction between the electrons with the same spin as within the B–H and N–H bonds). The repulsive contribution from the interaction between the lone electron pair of ammonia with the electrons of B–H bonds (Δρ Pauli1 ) is dominating in absolute terms; however, it does not influence the barrier. These results confirm the ‘classical’ view that the internal rotational barrier in ammonia borane can be understood solely in terms of NH↔HB steric (Pauli) effects, with minor participation stemming from the hyperconjugation (Fig. 2) and geometry distortion term. It must be further noted that we performed a detailed study of the changes in Δq Paulii values (based on various sets of molecules) and have found that differences in the second decimal place are quantitatively meaningful.

Dominant Pauli repulsion deformation density channels, Δρ Pauli1 , Δρ Pauli2 , Δρ Pauli3 , together with the corresponding charge estimations, Δq Pauli1 , Δq Pauli2 , Δq Pauli3 in the selected ammonia borane conformations. The blue/red contours correspond to accumulation/depletion of electron density. S Fully optimized staggered isomer, E s geom eclipsed structure in the staggered geometry, E fully optimized eclipsed isomer
Finally, we performed similar ETS-NOCV and Pauli repulsion analyses for ethane and found that CH↔HC Pauli (kinetic) repulsion channels are responsible for the rotation of methyl groups (see Supplementary electronic material); this is in line with the conclusions reached first by Bickelhaupt and subsequently by other researchers [42–46]. It is important at this point to cite another important work in the field explaining the origin of rotation in ethane. It is possible to decompose the barrier into changes in the kinetic (ΔT) and potential energy contributions: nuclei–-nuclei (ΔVnn), nuclei–-electrons (ΔVne) and electrons-electrons (ΔVee), as done by Bader and others for ethane [18]. Such an approach, while very physical and compelling, does not allow, for example, separate discussion of the role of the hyperconjugation effect, which is well rooted in chemistry. It was shown that rigid rotation S→ E s geom leads to a rise in all of the destabilizing terms (ΔT ∼ +9 kcal mol−1, ΔVnn+ΔVee ∼ +16 kcal mol−1), whereas the electron-nuclei stabilization is ΔVne ∼ −22 kcal mol−1. (Fig. 6.11 in [18]). At this non-equilibrium geometry, the rise in kinetic energy is related, through the virial theorem, to the repulsion force acting on the nuclei. Accordingly, in the next step, E s geom→E, the CC bond elongates, which leads to weakening of the attraction (ΔVne ∼ +207 kcal mol−1) and decrease in the repulsion (ΔVnn+ΔVee ∼ –201 kcal mol−1; ΔT ∼ –3 kcal mol−1, the values are provided with respect to ethane in staggered geometry). One should also note that various authors have combined the above contributions in different ways [31]. Finally, Liu and Govind [91], defined in an elegant way at DFT level, the steric contribution (equal to the kinetic Weizsäcker term) from a difference between the total electronic energy and the sum of electrostatic (ΔVne+ΔVee+ΔVnn) and quantum energy terms ΔEq (comprising the sum ΔExc+ΔEPauli); the change in the kinetic term due to the Pauli exclusion principle is incorporated in ΔE Pauli. It was shown that rigid rotation S→ E s geom results in the appearance of destabilizing forces originating from the fermionic quantum contribution ΔE q; closer inspection of the author’s data shows that this change is due entirely to a rise in the kinetic energy term [91]. Finally, one should cite the separate work of Nagy [92], who discussed the Fisher information based on the kinetic term; the role of kinetic energy and the information origin of the chemical bonding have been studied by Nalewajski [22].
Concluding remarks
The present work studied for the first time the internal rotation in ammonia borane based on our recently developed charge and energy decomposition scheme, ETS-NOCV, as well as the eigenvectors for Pauli repulsion. Detailed analyses of the electronic and the steric factors were performed in order to understand the origin of the barrier to rotation in ammonia borane.
We found that the barrier to rotation, staggered ↔ eclipsed, is only ∼2 kcal mol−1. It was demonstrated using the ETS-NOCV scheme that the hyperconjugation, originating from the charge transfer from the occupied σ (B–H) orbitals into the empty σ*(N–H), favors the staggered isomer, although, quantitatively it leads to only a slight stabilization (∼5 kcal mol−1). For ethane, this stabilization was more pronounced, ∼10 kcal mol−1. We have found, based on our newly proposed scheme, the natural orbitals for Pauli repulsion, that rigid rotation from the staggered to the eclipsed conformation causes predominantly the enhancement of steric (Pauli) repulsion acting solely between N–H and B–H bonds; this is subsequently ‘relieved’, leading to elongation of the B–N bond in the fully optimized eclipsed structure. Analogous trends were found for ethane (Table S1). Accordingly, the barrier to rotation in ammonia borane can be understood in a classical way; namely, as originating from the steric (Pauli) repulsion contributions that act solely between N–H and B–H bonds. Repulsion between the lone pair of ammonia and the B–H bonds is dominant in absolute terms; however, it does not influence the barrier.
References
Frenking G, Frohlich N (2000) Chem Rev 100:717–774 (and references therein)
Bickelhaupt FM, Baerends EJ (2000) In: Lipkowitz KB, Boyd DB (eds) Reviews in computational chemistry: Kohn-sham density functional theory: predicting and understanding chemistry, vol 15. Wiley, Hoboken, pp 1–86. doi: 10.1002/9780470125922.ch1
Hund F (1926) Z Phys 36:657–674
Mulliken RS (1927) Phys Rev 29:637–649
Boys SF (1960) Mol Phys 32:296–299
Edmiston C, Ruedenberg K (1963) Rev Mod Phys 35:457–464
Reed AE, Curtiss LA, Weinhold F (1998) Chem Rev 88:899–926
Wiberg K (1968) Tetrahedron 24:1083–1096
Gopinathan MS, Jug K (1983) Theor Chim Acta 63:497–509
Mayer I (1984) Chem Phys Lett 97:270–274
Nalewajski RF, Mrozek J (1994) Int J Quantum Chem 51:187–200
Nalewajski RF, Mrozek J, Formosinho SJ, Varandas AJC (1994) Int J Quantum Chem 52:1153–1176
Nalewajski RF, Mrozek J (1996) Int J Quantum Chem 57:377–389
Nalewajski RF, Mrozek J, Mazur G (1996) Can J Chem 74:1121–1130
Nalewajski RF, Mrozek J, Michalak A (1997) Int J Quantum Chem 61:589–601
Nalewajski RF, Mrozek J, Michalak A (1998) Polish J Chem 72:1779–1791
Michalak A, De Kock R, Ziegler T (2008) J Phys Chem A 112:7256–7263
Bader RF (1990) Atoms in molecules. A quantum theory. Oxford University Press, Oxford, and references therein
Ponec R (1997) J Math Chem 21:323–333
Jackobsen H (2010) Dalton Trans 39:5426–5428
Becke AD, Edgecombe KE (1990) J Chem Phys 92:5397–5403
Nalewajski RF (2010) Information origins of chemical bond, chemistry research and applications series. Nova Science, New York
Dapprich S, Frenking G (1995) J Phys Chem 99:9352–9362
Kitaura K, Morokuma K (1976) Int J Quantum Chem 10:325–340
Ziegler T, Rauk A (1977) Theor Chim Acta 46:1–10
Ziegler T, Rauk A (1979) Inorg Chem 18:1755–1759
Pendás AM, Blanco MA, Francisco E (2006) J Chem Phys 125:184112–184120
Jeziorski B, Moszynski R, Szalewicz K (1994) Chem Rev 94:1887–1930
Toro-Labbé A, Gutiérrez-Oliva S, Murray JS, Politzer P (2009) J Mol Model 15:707–710
Van Zeist WJ, Bickelhaupt FM (2010) Org Biomol Chem 8:3118–3127
Payne PW, Allen LC (1977) In: Schaefer HF (ed) Modern theoretical chemistry. Plenum, New York, pp 29–108
Wiberg KB (1998) Encyclopedia of computational chemistry, Schelyer PvR. Wiley, Berlin, pp 2518–2525
Kemp JD, Pitzer KS (1936) J Chem Phys 4:749
Sovers OJ, Kern CW, Pitzer RM, Karplus M (1968) J Chem Phys 49:2592–2599
Mulliken RS (1939) J Chem Phys 7:339–352
Lowe JP (1973) Science 179:527–532
England W, Gordon MS (1971) J Am Chem Soc 93:4649–4657
Epiotis ND, Cherry WR, Shaik S, Yates RL, Bernardi F (1977) Structural theory of organic chemistry. In: Topics in current chemistry, vol 70. Springer, Berlin
Bader RFW, Cheeseman JR, Laidig KE, Wiberg KB, Breneman C (1990) J Am Chem Soc 112:6530–6536
Pophristic V, Goodman L (2001) Nature 411:565–568
Weinhold F (2003) Angew Chem Int Ed 42:4188–4194
Bickelhaupt FM, Baerends EJ (2003) Angew Chem Int Ed 42:4183–4188
Pendás AM, Blanco MA, Francisco E (2009) J Comput Chem 30:98–109
Mo Y (2010) J Org Chem 75:2733–2736
Mo Y, Wu W, Song L, Lin M, Zhang Q, Gao J (2004) Angew Chem Int Ed 43:1986–1990
Mo Y, Gao Y (2007) Acc Chem Res 40:113–119
Mitoraj M, Michalak A, Ziegler T (2009) J Chem Theory Comput 5:962–975
Mitoraj M (2011) J Phys Chem A 115:14708–14716
Kurczab R, Mitoraj MP, Michalak A, Ziegler T (2010) J Phys Chem A 114:8581–8590
Mitoraj MP, Michalak A (2011) Inorg Chem 50:2168–2174
Michalak A, Mitoraj M, Ziegler T (2008) J Phys Chem A 112:1933–1939
Mitoraj M, Michalak A (2007) J Mol Model 13:347–355
Mitoraj MP, Zhu H, Michalak A, Ziegler T (2009) Int J Quantum Chem 109:3379–3386
Mitoraj M, Michalak A (2007) Organometallics 26:6576–6580
Mitoraj M, Michalak A (2008) J Mol Model 14:681–687
Srebro M, Mitoraj M (2009) Organometallics 28:3650–3655
Nalewajski RF (1993) KöSter AM, Jug K. Theor Chim Acta 85:463–484
Parafiniuk M, Mitoraj MP (2013) Organometallics 32:4103–4113
Dyduch k, Mitoraj MP, Michalak A (2013) J Mol Model 19:2747-2758.
Kozyra P, Brocławik E, Mitoraj MP, Datka J (2013) J Phys Chem 117:7511–7518
Mitoraj MP, Michalak A, Ziegler T (2009) Organometallics 28:3727–3733
Mitoraj MP, Parafiniuk M, Srebro M, Handzlik M, Buczek A, Michalak A (2011) J Mol Model 17:2337–2352
Piękoś Ł, Mitoraj MP (2013) J Comput Chem 34:294–304
Paul A, Musgrave CB (2007) Angew Chem 119:8301–8304
Baerends EJ, Autschbach J, Bashford D, Berger JA, Bérces A, Bickelhaupt FM, Bo C, de Boeij PL, Boerrigter PM, Cavallo L, Chong DP, Deng L, Dickson RM, Ellis DE, van Faassen M, Fan L, Fischer TH, Fonseca Guerra C, Giammona A, Ghysels A, van Gisbergen SJA, Götz AW, Groeneveld JA, Gritsenko OV, Grüning M, Harris FE, van den Hoek P, Jacob CR, Jacobsen H, Jensen L, Kadantsev ES, van Kessel G, Klooster R, Kootstra F, Krykunov MV, van Lenthe E, Louwen JN, McCormack DA, Michalak A, Mitoraj M, Neugebauer J, Nicu VP, Noodleman L, Osinga VP, Patchkovskii S, Philipsen PHT, Post D, Pye CC, Ravenek W, Rodriguez JI, Romaniello P, Ros P, Schipper PRT, Schreckenbach G, Seth M, Snijders JG, Solà M, Swart M, Swerhone D, te Velde G, Vernooijs P, Versluis L, Visscher L, Visser O, Wang F, Wesolowski TA, van Wezenbeek EM, Wiesenekker G, Wolff SK, Woo TK, Yakovlev AL, Ziegler T (2009) ADF 2009.01 SCM, Amsterdam, The Netherlands.
te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJA, Snijders JG, Ziegler T (2001) J Comput Chem 22:931–967, and references therein
Baerends EJ, Ellis DE, Ros P (1973) Chem Phys 2:41–51
Boerrigter PM, te Velde G, Baerends E (1988) J Int Quantum Chem 33:87–113
Becke A (1988) Phys Rev A 38:3098–3100
Perdew JP (1986) Phys Rev B 34:7406–7406
Visser O, Leyronnas P, van Zeist WJ, Lupki M (2009) ADF-GUI, SCM, Amsterdam, The Netherlands, http://www.scm.com.
Schwarz WHE, Schmidbaur H (2012) Chem Eur J 18: 4470–4479, and references therein
Ruedenberg K (1962) Rev Mod Phys 34:326–352
Tokiwa H, Ichikawa H (1994) Int J Quantum Chem 50:109–112
Pinter B, Fievez T, Bickelhaupt FM, Geerlings P, De Proft F (2012) Phys Chem 14:9846–9854
Haaland A (1989) Angew Chem Int Ed 101:1017–1032
Haaland A (1989) Angew Chem Int Ed 28:992–1007
Jonas V, Frenking G, Reetz MT (1994) J Am Chem Soc 116:8741–8753
Jagielska A, Moszynski R, Piela L (1999) J Chem Phys 110:947–954
Dixon DA, Gutowski M (2005) J Phys Chem A 109:5129–5135
Bauschlicher CW Jr, Ricca A (1995) Chem Phys Lett 237:14–19
Mo Y, Song L, Wu W, Zhang Q (2004) J Am Chem Soc 126:3974–3982
Plumley JA, Evanseck JD (2007) J Phys Chem A 111:13472–13483, and references therein
Bessac F, Frenking G (2006) Inorg Chem 45:6956–6964
Fernández I, Frenking G (2006) Chem Eur J 12:3617–3629
Thorne LR, Suenram RD, Lovas FJ (1982) J Chem Phys 78:167–171
Leboeuf M, Russo N, Salahub DR, Toscano M (1995) J Chem Phys 103:7408–7413
Poater J, Sola M, Bickelhaupt MF (2006) Chem Eur J 12:2889–2895
Goodman L, Hongbing G, Pophristic V (1999) J Chem Phys 110:4268–4275
Bader RFW, Preston HJT (1966) Can J Chem 44:131–145, and references therein.
Liu S, Govind N (2008) J Phys Chem A 112:6690–6699
Nagy Á (2007) Chem Phys Lett 449:212–215
Acknowledgments
M.P.M. greatly appreciates extremely valuable discussions with Dr. Chad McKee and Prof. von Rague Schleyer (University of Georgia) on the origin of rotational barriers in hydrocarbons. M.P.M. also acknowledges the financial support from the Polish Ministry of Science and Higher Education (“Outstanding Young Researchers” scholarships, 2010, 2011–2014 and for young researchers T-subsidy). M.Parafiniuk acknowledges the Polish Ministry of Science and Higher Education for the support of the project “Diamentowy Grant,” No. DI2011 010241. We are also grateful to Academic Computer Center CYFRONET (PL-Grid Infrastructure) for the computer time.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper belongs to Topical Collection MIB 2013 (Modeling Interactions in Biomolecules VI)
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 75 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Parafiniuk, M., Mitoraj, M.P. On the origin of internal rotation in ammonia borane. J Mol Model 20, 2272 (2014). https://doi.org/10.1007/s00894-014-2272-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2272-y