
Abstract
Objectives
Cell-free DNA (cfDNA) harboring mutations has been found in patients with diseases. Experimental studies have shown that cfDNA can be transmitted, leading to transformations in the host. In the present study, we evaluated whether bone allograft material contains cfDNA and whether this foreign cfDNA can be released into the patient’s blood circulation.
Materials and methods
Plasma samples were collected preoperatively and postoperatively on the same day, at 5 weeks, and 4 months from 25 women who received bone allograft material (test group) from male donors and from 10 women who were treated with autologous graft (control group, only pre- and postoperative samples were collected). DNA was quantified and characterized in bone material and plasma samples by quantitative PCR with primers specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Y chromosome and gel electrophoresis. DNA in bone material was digested by different concentrations of DNase I.
Results
We detected between 1 and 1.8 μg cfDNA fragments at a length around 601 base pairs (bp) and smaller in each 100 mg allograft. Treatment of the allograft with DNase I completely degraded the longer but not the shorter DNA 90-bp fragments. Y-DNA was not detected in the patients’ bloodstream at any time during the treatment and follow-up, but elevated levels of circulating cfDNA could be measured immediately postoperatively.
Conclusions
Our results suggest that a transmission of DNA from allografts used for alveolar ridge reconstruction in humans is unlikely. The observed increase in circulating cfDNA in allograft and autograft patients immediately postoperatively may be elicited by the surgical procedure.
Clinical relevance
The results support the safety of allograft materials. The results suggest that human allograft materials seem not to release DNA into the blood since we did not measure Y-DNA with our technique.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A reduced alveolar bone volume is a physiological consequence after tooth loss, commonly implying the need for augmentation procedures prior to dental implant treatment. In current scientific literature, autogenous bone is still considered the gold standard for augmentation procedures in reconstructive surgery and dental implantology [1,2,3], due to its osteogenic, osteoinductive, and osteoconductive properties including lack of immunogenicity. However, autogenous bone grafts may show different disadvantages, such as increased operation time, donor site morbidity, post-operative discomfort, limitations in bone quantity and volume, unpredictable bone quality, and reduced volume stability, as well as a fast resorption rate. It may also be only effective under good recipient site conditions [2, 4,5,6,7,8].
Preferred donor regions for autologous bone harvesting are extraoral donor sites like the iliac crest as well as intraoral donor sites, preferably from the interforaminal region and the retromolar area of the mandible [9]. Based on current insights from clinical human studies, bone grafts from the iliac crest are suitable for the reconstruction of large defects and atrophic edentulous alveolar crests in the mandible and maxilla [10,11,12]. Despite the still widespread use of iliac bone grafts for reconstructive surgery in dentistry, harvesting—especially from the anterior part of the iliac crest—partially leads to a prolonged operating time as well as to major complications and donor site morbidity, including extended healing periods, chronic donor site pain, functional impairment, wound dehiscence, and deep infections [9, 13,14,15,16,17]. Intraoral donor regions for autologous bone block harvesting have shown advantages in avoiding the aforementioned drawbacks of bone harvesting from the iliac region, leading to good clinical results and minor complications [18]. However, a further disadvantage of autologous bone blocks may be the lacking possibility to create individual transplants being defect customized by using CAD-CAM technology. Nonetheless, intraoral bone harvesting also produces donor site morbidity, which is more pronounced in the interforaminal region of the chin [9]. Furthermore, the availability of autologous bone is limited. In order to overcome this limitation on one hand and to avoid patient discomfort on the other hand, efforts were made to find suitable alternatives for bone replacement, particularly when grafts are required for large intraoral defects [3]. Among those materials, bone allografts are considered a very useful alternative because they provide similar properties like autogenous bone but are available in unlimited volume. As allogeneic grafts comprise risks of disease transmission—in particular human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV)—standardized sterilization procedures and disease testing are important issues for patient safety [3, 19]. Application of different techniques to control infection transmission, such as donor screening, repeated infectious disease testing, graft sterilization with ethylene oxide or radiation, and long-term tracking of the grafts, significantly decreases the disease transmission risks [3, 20].
Maxgraft® is an allogeneic bone replacement material, grafted from femoral heads of living donors and available as granules or bone blocks. Donor bone is treated in a validated multi-step chemical cleaning process, which prevents disease transmission by the removal of cells, viruses, antigens, and pathogens [21, 22]. This process comprises delipidation, osmotic and oxidative treatment, solvent dehydration, and sterilization through limited-dose gamma radiation [23]. The trabecular matrix of bone allograft materials promotes the regeneration and assimilation of the recipient tissue and, in particular, the bone remodeling process, which is nearly completed by the transformation of the material in native bone within a few months.
However, using allogeneic bone material prior to implantation of dental implants can increase the risk of complications. In some ways, the material is not biocompatible. Although allografts might have osteoinductive and osteoconductive functions, those properties are still discussed contradictorily [4, 7, 24,25,26]. Demineralized freeze dried bone allograft (DFDBA) has been shown to be osteoconductive and osteoinductive due to the release of BMPs [27], although clinical outcomes comparing FDBA and DFDBA have been reported to be similar [28]. In vitro and animal investigations have revealed osteoinductive functions of DFDBA by recruiting cells and ectopic bone formation [29]. Other authors discussed the lack of osteoinductive capacities. Disadvantages of allogeneic materials may be a protracted vascularization, slow remodeling and resorption, or longer time for osseointegration as well as the risk of immunogenic reactions.
One of the goals of this project was to investigate whether the allogeneic bone materials contain cell-free DNA (cfDNA) from the donor. Therefore, we included only female patients in this study and used allograft materials only from male donors in order to test whether there is Y-cfDNA in the plasma of female patients after implantation. Our hypothesis was that this foreign DNA could enter the blood circulation of patients who had received intraoral bone augmentation prior to implantation with dental implants, thereby increasing the risk of disease transmission (from the donor to the patient).
Materials and methods
Study design and participants
In this prospective cohort study, 35 systemically healthy, partially edentulous female patients with alveolar ridge defects and the need for implant treatment were treated in a single dental office. Lateral augmentation procedures of the alveolar ridge were performed in a two-stage surgery with a particulate allograft material and collagen membranes. Maxgraft® allograft spongiosa particle (Botiss Company, Berlin, Germany, part of Straumann Group, Basel, Switzerland) was used in 25 individuals of the test group, whereas autogenous bone, harvested intraorally, was used for guided bone regeneration in 10 individuals as controls.
We classified alveolar ridge defects according to the description published by Seibert in 1983 [30], who differentiated between three defect classes: buccolingual vertical (class I), apicocoronal (class II), and combined defects (class III). Here, only patients with Seibert class I or II defects were included.
All study procedures were performed in compliance with the Helsinki declaration and were approved by an ethics committee (Ethik-Kommission der Ärztekammer Hamburg, Hamburg, Germany, PV5211) as well as by the German Registry of Clinical Studies (DRKS 00013010). All blood samples were consistently processed and managed as described in specific protocols, no tissue biopsies needed to be harvested for our investigations. The sample flow is visualized in Fig. 1.

Sample flow and study procedure. The flow-chart shows the initial investigation of the different charges of the bone allograft material as well as the four different time points for blood sample collection and the subsequent laboratory experiments. Furthermore, the clinical procedure for the test and the control groups are shown
Surgical procedure
After written informed consent of each patient was obtained, grafting of the alveolar ridge and subsequent implant placement were performed under local anesthesia with Ultracain-DS forte (Sanofi-Aventis, Frankfurt/Main, Germany). Before surgery, venous blood samples for DNA analyses were collected from patients. After deflection of a mucoperiosteal flap, a cortical perforation was created, and the bone grafting material was inserted. The allograft was covered with the respective collagen membrane (Jason Membrane, Botiss Company) for guided tissue regeneration. Thereafter, the periosteum of the mucoperiosteal flap was cut in order to mobilize the flap for a tension-free closure of the surgical site. Flap-fixation was performed using a horizontal and vertical mattress suture with 5.0 Goretex filaments (W. L. Gore & Associates GmbH, Putzbrunn, Germany) (Fig. 2a–d). A 2.0% chlorhexidine rinsing solution was administered for postoperative oral hygiene. Immediately after surgery, another venous blood sample was taken for DNA analyses. Postoperative appointments were scheduled after 1, 2, 6, and 12 weeks. Sutures were removed 2 weeks after augmentation.

Overview of the surgical procedure for bone augmentation. Following reflection of a mucoperiosteal flap, an extremely thin alveolar crest was visible (a). Subsequently, the ridge was prepared with cortical perforations. The bone allograft material, soaked in the 2nd phase of the PRGF solution, was used to build up the alveolar bone to the necessary volume for future implant placement (b) and covered with a resorbable collagen membrane (c). Using horizontal mattress sutures (c) and a continuous half-hitch suture (d), primary closure was achieved. Approximately 4 months later, the second surgical procedure was performed. A mucoperiosteal flap was raised and the substantial gain of alveolar bone volume could be appreciated (e). Three osteotomies were performed according to the manufacturer’s recommendations (ASTRA Dentsply) (f) and three implants were inserted (g). The postoperative panoramic radiograph demonstrated the placement of three implants of an adequate dimension (h), distributed as preoperatively planned. The placement of implants of such a dimension would have been impossible without bone augmentation beforehand, PRGF, plasma rich in growth factors
Implants were inserted after a mean healing period of 5.4 months after bone augmentation in an open flap approach. Implants (Straumann Group; Camlog GmbH, Wimsheim, Germany; Astra Implant System, Dentsply Sirona Implants, Mannheim, Germany) with a diameter of at least 3.3 mm and a minimum length of 8.0 mm were inserted with a mean insertion torque of 35 Ncm according to the manufacturer’s recommendations. After implant placement, the mucoperiosteal flap was readapted and fixed with Gore-Tex sutures and a postoperative radiograph was taken (Fig. 2e–h). Uncovering of the implants and prosthetic restoration of implants proceeded after a mean submerged healing period of 4 months.
All treatments were provided by a single surgeon (ÖS) within a specialty practice limited to periodontology and implant dentistry in Hamburg, Germany. In this specialty practice, the patients were treated in accordance with established guidelines.
Collection of plasma samples
Blood samples were collected from 25 women who received bone allograft material from male donors (allograft patients) and from 10 women who were treated with autologous graft (autograft patients). Blood samples of the patients were obtained between September 2016 and February 2018. Blood samples were collected on the day of surgery preoperatively, immediately postoperatively, and at 5 weeks and 4 months after surgery. The mean age of allograft patients was 58 years (range 39–78) and that of autograft patients was 55 years (range 32–76).
DNA digestion of bone allograft material
Hundred-milligram bone allograft material of eight different charges of bone allograft material from male donors (maxgraft®, Straumann Group) was digested in 400 μL reaction buffer (total volume) with 1 μL (50 units), 2 μL (100 units), or 5 μL (250 units) DNase I (Thermo Fisher Scientific, Darmstadt, Germany) and incubated at 37 °C and 1000 rpm on a thermomixer (Eppendorf, Hamburg, Germany) for 30 min.
DNA extraction from bone allograft material
Undigested and digested DNA was extracted from the untreated and treated bone material pellet, respectively, using the QIAamp DNA Investigator Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Briefly, the pellets were dissolved in 360 μL ATL buffer plus 20 μL of proteinase K and incubated at 56 °C overnight. Next day, 300 μL AL buffer was added to the reaction. After incubation at 70 °C and 900 rpm for 10 min, the supernatant mixed with 700 μL 100% ethanol was applied on a QIAmp MinElute column. After washing the column with 600 μL AW1 buffer, 700 μL AW2 buffer, and 700 μL 100% ethanol, the DNA was precipitated from the column by 30 μL nuclease-free water.
Quantity of the extracted cfDNA was determined spectrophotometrically using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). Briefly, 2 μL DNA was diluted in 198 μL HS Reagent solved in HS Buffer (1:200), incubated for 3 min at room temperature and measured on the Qubit 2.0 Fluorometer (Thermo Fisher Scientific). The fluorometer was calibrated with standard 1 and 2 of the kit. We repeated the measurement of each sample once and got the same data.
PCR
Two microliters of undigested and digested DNA derived from bone allograft material was amplified with 0.5 μL 10 μM of 4 different primer pairs specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Supplemental Table 1 specifying the primer sequence, the annealing temperatures, and elongation times) and 10 μL Taq PCR Master Mix (Qiagen). The reactions were run on an MJ Research PTC-200 Peltier Thermal Cycler (Global Medical Instrumentation, Ramsey, MN, USA): 1 cycle at 95 °C for 5 min; 40 cycles at 95 °C for 30 s, 55–60 °C for 30 s, and 72 °C for 30–45 s; and 1 cycle at 72 °C for 5 min. A negative control without any templates was also included. The PCR products were analyzed on a 1.5% agarose gel (Biozym Scientific, Hessisch Oldendorf, Germany).
Verification of hemolysis in plasma samples
EDTA blood was collected by PAXgene Blood DNA Tubes (Qiagen). Plasma was prepared by two centrifugation steps at 3000g and 13,000g for 10 min. Following centrifugation, the supernatant contained plasma.
To avoid quantifying circulating DNA in hemolytic plasma samples that may influence our results, we performed hemoglobin measurements by spectral analysis. Red blood cells were lysed in 7 mL whole blood by erythrocyte lysis buffer (0.3 M sucrose, 10 mM Tris pH 7.5, 5 mM MgCl2, 1% Triton X100). A dilution series (1:1, 1:3, 1:4, 1:6, 1:8, 1:10, 1:12, 1:14, 1:18, 1:20) of lysed red blood cells was prepared in plasma and served as a standard curve for the measurement of hemolysis in all plasma samples. Fifty microliters of each plasma sample was measured in duplicate on a microplate reader (Tecan, Männerdorf, Switzerland). Absorbance peaks at 414, 541, and 576 nm indicated free hemoglobin, with the highest peak at 414 nm. The higher the absorbance in plasma samples is the higher the degree of hemolysis is. The average values and standard deviations were calculated from the duplicate (Fig. 3).

Levels of free hemoglobin measured in plasma samples, Hemolysis was assessed by spectrophotometry at wavelengths from 350 to 650 nm (a). A dilution series of lysed red blood cells in plasma was prepared (b). The degree of hemolysis was determined based on the optical density (OD) at 414 nm (absorbance peak of free hemoglobin, called Soret band), with additional peaks at 541 and 576 nm (a). Samples were classified as being hemolyzed if the OD at 414 nm exceeded 0.25. The integrated scatter plot of plasma samples comprised values from 0.01 to 0.50. Plasma samples with a value above 0.25 (red line) were lysed controls and not analyzed (c)
Extraction of circulating cfDNA from plasma samples
Using PAXgene Blood ccf Tubes (Qiagen), blood was collected preoperatively and postoperatively (collected on the same day) from 25 allograft patients and 10 autograft patients. Additionally, blood was withdrawn from allograft patients, 5 weeks and 4 months after surgery. Plasma was prepared by two centrifugation steps at 2671 and 10,000g, each for 10 min.
Circulating cfDNA was extracted from a total of 120 plasma samples using the QIAamp Circulating Nucleic Acid Kit (Qiagen) according to the manufacturer’s instructions. Briefly, 3 mL plasma was mixed with 2.4 mL ACL buffer (containing 0.2 μg/μL carrier RNA) plus 300 μL of proteinase K for 30 s, and incubated at 60 °C for 30 min. After addition of 5.4 mL ACB to the reaction and incubation on ice for 5 min, the lysate was drawn through a QIAmp Mini column. The column was washed with 600 μL ACW1 buffer, 750 μL ACW2 buffer, and 700 μL 100% ethanol using vacuum of six bar. The DNA was precipitated from the column by 30 μL AVE buffer. Quantity of the extracted circulating DNA was determined spectrophotometrically using the Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific), as described above.
Real-time PCR to detect the Y chromosome
First, 6.25 μL circulating DNA was pre-amplified using 12.5 μL TaqMan Preamp Master Mix and 6.25 μL of a primer pool containing primers for GAPDH (housekeeping gene as control) and the sex-determining region of the Y chromosome (SRY), diluted 1:100 in Tris-EDTA (Thermo Fisher). The reactions were run on an MJ Research PTC-200 Peltier Thermal Cycler (Global Medical Instrumentation): 1 cycle at 95 °C for 10 min and 15 cycles at 95 °C for 15 s and at 60 °C for 4 min. Then, 2.5 μL pre-amplified cfDNA was amplified using 5 μL TaqMan Expression Master Mix and 0.5 μL primers GAPDH and SRY (Thermo Fisher). The reactions were again run on an MJ Research PTC-200 Peltier Thermal Cycler (Global Medical Instrumentation): 1 cycle at 95 °C for 10 min and 40 cycles at 95 °C for 15 s and at 60 °C for 1 min. A negative control without any templates and a positive control, cfDNA from a male, were also included.
Statistical analysis
Statistical analyses were performed using the SPSS software package, version 22.0 (SPSS Inc. Chicago, IL, US). Differences in group levels for nonparametric comparisons were bivariately assessed by univariate analyses of the Mann-Whitney U test of two independent variables and the Wilcoxon test of two dependent variables. All p values are two-sided. Due to the explorative nature of the study, no formal adjustment for multiple testing was performed.
Results
Determination of DNA amounts and size in bone allograft material
At first, we analyzed eight batches of male bone allograft material, whether they contain cfDNA from the donor that could affect patient morbidity. Table 1 summarizes the spectrophotometric DNA measurements performed with the Qubit dsDNA HS Assay Kit. Although the bone allograft material was treated with UV light, we detected DNA ranging from 1 to 1.8 μg in 100 mg of the samples. The lengths of the DNA fragments from one batch were determined by PCR using four primer sets specific for different sequences within the household gene GAPDH. The primer pairs generated PCR products at sizes of 90, 201, 601, and 877 base pairs (bp) (Fig. 4a–c; lanes 1). However, primers that should amplify a GAPDH sequence at a size of 994 bp and larger did not produce any PCR product since real-time PCR (without the use of a TaqMan probe) is not qualified for such amplifications (data not shown). Taken together, gel electrophoresis of the PCR products showed that the bone allograft material contained DNA fragments at a length of 877 bp and smaller (Fig. 4a–c; lanes 1), and probably longer, as observed by the analysis of the whole unamplified DNA showing a smear on the gel (data not shown).

Characterization of donor cfDNA in bone allograft material. The lengths of undigested DNA and DNA digested by different concentrations of DNase I in bone allograft material were determined by gel electrophoresis (1.5% agarose) of real-time PCR products encompassing different sizes and locations of the housekeeping gene GAPDH. Lane M, DNA marker; lane 1, undigested DNA; lane 2, DNA digested with 50 U DNase I; lane 3, DNA digested with 100 U DNase I; lane 4, DNA digested with 250 U DNase I; and lane C, control without DNA (a, b, c)
We digested DNA directly in one batch of bone grafting material with various concentrations (50, 100, 250 U) of DNase I. Concentrations of 100 and 250 units of DNase I completely degraded the longer DNA fragments at sizes of 301–877 bp but not the shorter 90-bp DNA fragments (Fig. 4a–c; lanes 2–4).
As shown in Table 1, we also measured Y-DNA in the eight batches of male bone allograft material using TaqMan real-time PCR with SRY primers. The size of the measured relative Ct values did not correlate with the contents of total DNA, indicating that higher cell-free DNA amounts do not necessarily contain higher Y-DNA amounts, and thus, the proportion of Y-DNA in total DNA may vary (Table 1).
Characterization of circulating DNA in plasma
At first, we examined the plasma samples for hemolysis. None of the 120 plasma samples were hemolyzed, and all samples could be analyzed (Fig. 3). To examine whether foreign DNA from the allogeneic bone material migrates into the patients’ blood circulation, plasma samples from the female allograft patients who received male donors’ material were analyzed. Circulating cfDNA could have been recognized as foreign if these women harbored Y-cfDNA fragments in their bloodstream. Therefore, the above real-time PCR with SRY primers was also carried out on plasma samples taken at different times from the allograft patients (one before surgery, one on the same day after surgery, one 5 weeks later, and one 4 months later). Pre- and postsurgical plasma samples (same day) from the 10 autograft patients as well as from an untreated woman served as negative controls, whereas a plasma from a man served as a positive control. At no point in time, Y-cfDNA was detected in the allograft patients’ plasma samples, indicating that no male DNA from the allogeneic bone material migrated into the patients’ bloodstream (Fig. 5).

Determination of Y-cfDNA fragments in plasma. The amplification curves below the threshold (red line) do not provide any PCR product of the Y chromosome in plasma samples derived from women undergoing intraoral bone augmentation with allogeneic material and display background. The amplification curves above the threshold show successful amplification of male cfDNA derived from the allogeneic bone material and served as a positive control
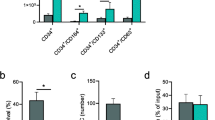
Thereafter, we measured the levels of circulating DNA in the patients’ plasma to determine whether the surgical intervention itself causes the release of DNA into the blood by the body’s own dying (apoptotic) cells. As shown in Fig. 6, the levels of circulating plasma cfDNA were higher in both, allograft and autograft patients, directly after surgery (p = 0.001). In addition, circulating DNA started to decrease slowly in allograft patients 5 weeks and 4 months after surgery, although the differences were not statistically significant (Fig. 6). A statistical comparison showed that the levels of circulating plasma cfDNA were independent of the patient’s age.

cfDNA levels in plasma of allograft and autograft patients. The box blot shows the levels of circulating cfDNA measured in plasma of 25 women undergoing intraoral bone augmentation with allogeneic material (on the left) before and after surgery. Additionally, pre- and postsurgical plasma samples from 10 patients undergoing intraoral bone augmentation with autologous material (on the right) were assessed. Differences between presurgical samples and the first postsurgical samples were statistically significant (p = 0.001) for both cohorts. Before surgery; 1st after, same day after surgery; 2nd after, 5 weeks after surgery; 3rd after, 4 months after surgery
Finally, the autograft group harbored a much larger range of the cfDNA concentrations in their plasma than the allograft group. However, the differences in cfDNA levels between the allograft and autograft group were not significant before surgery (p = 0.361) and after surgery (on the same day, p = 0.086).
Discussion
In the current study, we characterized cfDNA in bone allograft material from male donors as well as in plasma samples from women who received either the aforementioned allograft material or an autologous graft. Although the patient cohort of 25 women and reference cohort of 10 women seem to be small, our analyses show robust data. It should be considered that the samples were collected in a single-center approach by one clinic only, in order to allow a better comparability of the data.
In bone, there is barely information about the character and fate of cfDNA [31, 32]. This area of cfDNA physiology remains unclear and needs further examination. However, more is known on cfDNA in the blood circulation. In human blood, cfDNA circulates predominantly as nucleosomes [33]. On average, the size of cfDNA varies between small fragments of about 200 bp and large fragments as long as 21 kb. The size of cfDNA may indicate its source. Apoptotic cells produce DNA fragments of 180–200 bp or multiples of this unit, whereas necrotic cells release higher molecular-weight DNA fragments in size of over 10,000 bp [34]. Necrotic and apoptotic cells are usually phagocytosed by macrophages or other scavenger cells. CfDNA fragments can also be actively released. The levels of cfDNA have a great dynamic range in human blood and are influenced by physiological and pathological factors. The amounts of cfDNA are also influenced by clearance, degradation, and other physiological filtering events of the blood and lymphatic circulation. The nuclease activity in blood may also be a further important factor for the turnover of cfDNA. Typically, cfDNA is cleared from the blood by the liver and kidney and has a variable half-life in the blood circulation ranging from 15 min to several hours. Some forms of cfDNA may survive longer than others [35]. Considering its quality, the different cfDNA fragment lengths have important implications in the measurement and analysis of cfDNA.
Due to our unique study design, we were able to show for the first time data derived from three to four follow-up samples after surgery in these patients. The most important result of our study is that we did not detect Y-cfDNA in any of the postsurgical plasma samples from the female allograft patients, although the allogeneic bone material used for alveolar ridge reconstruction still contained amounts of cfDNA. This finding strongly suggests that no donor cfDNA migrated into the patients’ blood circulation, and supports the supposed safety of bone materials in terms of the unlikelihood of the potential transmission of diseases.
In line with our findings, small DNA amounts have also been found by other authors in allograft materials commonly used in dentistry [32, 36]. Using the NanoDrop 1000 spectrophotometer, Fretwurst et al. [32] measured a cfDNA concentration of 25.7 ng/mL in maxgraft®, an allograft that we also used. We detected cfDNA ranging from 1 to 1.8 μg in 100 mg of eight samples, referring to an average value of 14.2 ng/mg (or ng/mL) of this allograft material. Thus, the concentrations measured by us were approximately half as high as those measured by Fretwurst et al. This can be explained by the fact that we measured the cfDNA concentrations with the Qubit system, which is more precise in DNA measurements than the NanoDrop device, and that we used different charges. Performing digestion of this DNA, we were able to completely degrade the longer cfDNA fragments but not the shorter 90-bp cfDNA fragments. Possibly, this DNA can elicit an immune response leading to inflammatory processes. In this regard, it was reported that microbial/cytosolic nucleic acids induce potent innate immune responses by stimulating the expression of type I interferons [37]. However, bone material itself has a very low antigenic potential, which is in contrast to blood cells and endothelial cells. The dental allograft products are almost completely decellularized, and soluble proteins or DNA are only present in traces. Collagen in bone, on the other hand, is explicitly desired because it gives the material mechanical strength [38, 39].
We also detected significantly increased levels of circulating cfDNA in all patients after surgery compared with those before surgery. Probably, this DNA is released into the blood by dying cells, e.g., by apoptosis and necrosis or even actively by living cells. These increased post-surgical levels of circulating cfDNA in the blood of these patients could reflect inflammatory processes [40]. Although the range of the cfDNA concentrations was much larger in the plasma of the autograft than in the allograft group, the differences of cfDNA levels between the allograft and autograft group were not significant before surgery and after surgery. Why the autograft group harbors a heterogeneous cfDNA distribution cannot be explained with the analysis methods used in this study. Most likely, immunological reactions in the patients as well as surgical trauma caused by the harvesting of different amounts of the autogenous bone might have caused this heterogeneous cfDNA distribution. However, this is only speculative and should be further investigated in future studies. In both of our patient cohorts, remodeling processes during the healing of the substitutes including osteogenesis, angiogenesis, or resorption may induce apoptosis and possibly necrosis, which leads to the shedding of DNA into the bloodstream. In a histological study of allogenous bone graft remodeling in the mandible of rabbits, necrotic bone areas were observed during osteogenesis [41]. However, it is not well understood to what extent inflammatory reactions, e.g., sterile inflammation with cytokine upregulation and macrophage activation, may be a transient phenomenon in “physiological” healing of bone substitutes [42]. Further studies on the healing of allogeneic bone substitutes in patients are needed to test whether apoptosis and necrosis occur. Nevertheless, we cannot fully exclude the transmission of smaller cfDNA fragments from the donor material to the recipients which could not be detected by our methods, but to our knowledge, the only cases of alloimmunization from bone material reported to date occurred when massive fresh frozen material or osteochondral allograft was used [43, 44]. Although sensitization did not interfere with graft incorporation in the described reports, it may bear a potential risk to individual patients if they would require organ transplantation at some future time [44]. In addition, Fretwurst et al. [45] recently described the presence of major histocompatibility complex (MHC) molecules in protein extracts from processed allogeneic bone blocks, suggesting a potential antigenicity of allografts despite thorough processing.
In summary, we conclude that the residual risk of alloimmunization is low when allogeneic bone grafting material produced in accordance with current standards is used. In order to finally investigate the possibility of alloimmunization, additional studies will be carried out. These analyses could, for example, examine human biopsies and include other biological and immunological methods, such as basophil granulation and lymphocyte transformation assays, as well as the examination for MHC molecules.
References
Baj A, Trapella G, Lauritano D, Candotto V, Mancini GE, Giannì AB (2016) An overview on bone reconstruction of atrophic maxilla: success parameters and critical issues. J Biol Regul Homeost Agents 30:209–215
Sakkas A, Wilde F, Heufelder M, Winter K, Schramm A (2017) Autogenous bone grafts in oral implantology-is it still a “gold standard”? A consecutive review of 279 patients with 456 clinical procedures. Int J Implant Dent 3:23. https://doi.org/10.1186/s40729-017-0084-4
Sutherland D, Bostrom M (2005) Grafts and bone graft substitutes. In: Bone regeneration and repair. Springer, pp 133–156
Jamjoom A, Cohen RE (2015) Grafts for ridge preservation. J Funct Biomater 6:833–848. https://doi.org/10.3390/jfb6030833
Rogers GF, Greene AK (2012) Autogenous bone graft: basic science and clinical implications. J Craniofac Surg 23:323–327. https://doi.org/10.1097/SCS.0b013e318241dcba
Sanz M, Vignoletti F (2015) Key aspects on the use of bone substitutes for bone regeneration of edentulous ridges. Dent Mater 31:640–647. https://doi.org/10.1016/j.dental.2015.03.005
Wang W, Yeung KWK (2017) Bone grafts and biomaterials substitutes for bone defect repair: a review. Bioact Mater 2:224–247. https://doi.org/10.1016/j.bioactmat.2017.05.007
Yamada M, Egusa H (2018) Current bone substitutes for implant dentistry. J Prosthodont Res 62:152–161. https://doi.org/10.1016/j.jpor.2017.08.010
Nkenke E, Neukam FW (2014) Autogenous bone harvesting and grafting in advanced jaw resorption: morbidity, resorption and implant survival. Eur J Oral Implantol 7(Suppl 2):S203–S217
Fretwurst T, Nack C, Al-Ghrairi M et al (2015) Long-term retrospective evaluation of the peri-implant bone level in onlay grafted patients with iliac bone from the anterior superior iliac crest. J Craniomaxillofac Surg 43:956–960. https://doi.org/10.1016/j.jcms.2015.03.037
Lonie S, Herle P, Paddle A, Pradhan N, Birch T, Shayan R (2016) Mandibular reconstruction: meta-analysis of iliac- versus fibula-free flaps. ANZ J Surg 86:337–342. https://doi.org/10.1111/ans.13274
Schaudy C, Vinzenz K (2014) Osteoplastic reconstruction of severely resorbed maxilla by stack plasty: combining sinus augmentation with lateral and vertical onlay bone grafting. Br J Oral Maxillofac Surg 52:647–651. https://doi.org/10.1016/j.bjoms.2014.04.018
Arrington ED, Smith WJ, Chambers HG, Bucknell AL, Davino NA (1996) Complications of iliac crest bone graft harvesting. Clin Orthop Relat Res 329:300–309
Barone A, Ricci M, Mangano F, Covani U (2011) Morbidity associated with iliac crest harvesting in the treatment of maxillary and mandibular atrophies: a 10-year analysis. J Oral Maxillofac Surg 69:2298–2304. https://doi.org/10.1016/j.joms.2011.01.014
Dimitriou R, Mataliotakis GI, Angoules AG, Kanakaris NK, Giannoudis PV (2011) Complications following autologous bone graft harvesting from the iliac crest and using the RIA: a systematic review. Injury 42(Suppl 2):S3–S15. https://doi.org/10.1016/j.injury.2011.06.015
Pappalardo S, Puzzo S, Carlino V, Cappello V (2007) Bone substitutes in oral surgery. Minerva Stomatol 56:541–557
Silber JS, Anderson DG, Daffner SD, Brislin BT, Leland JM, Hilibrand AS, Vaccaro AR, Albert TJ (2003) Donor site morbidity after anterior iliac crest bone harvest for single-level anterior cervical discectomy and fusion. Spine (Phila Pa 1976) 28:134–139. https://doi.org/10.1097/01.BRS.0000041587.55176.67
Cordaro L, Torsello F, Accorsi Ribeiro C, Liberatore M, Mirisola di Torresanto V (2010) Inlay-onlay grafting for three-dimensional reconstruction of the posterior atrophic maxilla with mandibular bone. Int J Oral Maxillofac Surg 39:350–357. https://doi.org/10.1016/j.ijom.2010.02.016
Kolk A, Handschel J, Drescher W, Rothamel D, Kloss F, Blessmann M, Heiland M, Wolff KD, Smeets R (2012) Current trends and future perspectives of bone substitute materials - from space holders to innovative biomaterials. J Craniomaxillofac Surg 40:706–718. https://doi.org/10.1016/j.jcms.2012.01.002
Holtzclaw D, Toscano N, Eisenlohr L, Callan D (2008) The safety of bone allografts used in dentistry: a review. J Am Dent Assoc 139:1192–1199
Ramachandra SS, Rana R, Reetika S, Jithendra KD (2014) Options to avoid the second surgical site: a review of literature. Cell Tissue Bank 15:297–305. https://doi.org/10.1007/s10561-013-9395-8
Schoepf C (2006) Allograft safety: efficacy of the Tutoplast® process. Implantology 1:10–15
Feuille F, Knapp CI, Brunsvold MA, Mellonig JT (2003) Clinical and histologic evaluation of bone-replacement grafts in the treatment of localized alveolar ridge defects. Part 1: mineralized freeze-dried bone allograft. Int J Periodontics Restorative Dent 23:29–35
Garcia-Gareta E, Coathup MJ, Blunn GW (2015) Osteoinduction of bone grafting materials for bone repair and regeneration. Bone 81:112–121. https://doi.org/10.1016/j.bone.2015.07.007
Troeltzsch M, Troeltzsch M, Kauffmann P, Gruber R, Brockmeyer P, Moser N, Rau A, Schliephake H (2016) Clinical efficacy of grafting materials in alveolar ridge augmentation: a systematic review. J Craniomaxillofac Surg 44:1618–1629. https://doi.org/10.1016/j.jcms.2016.07.028
Danesh-Sani SA, Engebretson SP, Janal MN (2017) Histomorphometric results of different grafting materials and effect of healing time on bone maturation after sinus floor augmentation: a systematic review and meta-analysis. J Periodontal Res 52:301–312. https://doi.org/10.1111/jre.12402
Monje A, O’Valle F, Monje-Gil F, Ortega-Oller I, Mesa F, Wang HL, Galindo-Moreno P (2017) Cellular, vascular, and histomorphometric outcomes of solvent-dehydrated vs freeze-dried allogeneic graft for maxillary sinus augmentation: a randomized case series. Int J Oral Maxillofac Implants 32:121–127. https://doi.org/10.11607/jomi.4801
Wood RA, Mealey BL (2012) Histologic comparison of healing after tooth extraction with ridge preservation using mineralized versus demineralized freeze-dried bone allograft. J Periodontol 83:329–336. https://doi.org/10.1902/jop.2011.110270
Miron RJ, Sculean A, Shuang Y, Bosshardt DD, Gruber R, Buser D, Chandad F, Zhang Y (2016) Osteoinductive potential of a novel biphasic calcium phosphate bone graft in comparison with autographs, xenografts, and DFDBA. Clin Oral Implants Res 27:668–675. https://doi.org/10.1111/clr.12647
Seibert JS (1983) Reconstruction of deformed, partially edentulous ridges, using full thickness onlay grafts. Part II. Prosthetic/periodontal interrelationships. Compend Contin Educ Dent 4:549–562
Schwarzenbach H, Chun FK-H, Isbarn H, Huland H, Pantel K (2011) Genomic profiling of cell-free DNA in blood and bone marrow of prostate cancer patients. J Cancer Res Clin Oncol 137:811–819. https://doi.org/10.1007/s00432-010-0941-5
Fretwurst T, Spanou A, Nelson K, Wein M, Steinberg T, Stricker A (2014) Comparison of four different allogeneic bone grafts for alveolar ridge reconstruction: a preliminary histologic and biochemical analysis. Oral Surg Oral Med Oral Pathol Oral Radiol 118:424–431. https://doi.org/10.1016/j.oooo.2014.05.020
Roth C, Pantel K, Muller V et al (2011) Apoptosis-related deregulation of proteolytic activities and high serum levels of circulating nucleosomes and DNA in blood correlate with breast cancer progression. BMC Cancer 11:4. https://doi.org/10.1186/1471-2407-11-4
Grunt M, Hillebrand T, Schwarzenbach H (2017) Clinical relevance of size selection of circulating DNA. Transl Cancer Res 7:S171–S184
Schwarzenbach H, Hoon DSB, Pantel K (2011) Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 11:426–437. https://doi.org/10.1038/nrc3066
Fretwurst T, Nelson K, Schmelzeisen R, Stricker A (2014) Allogenes Knochenersatzmaterial: Sicheres Augmentat oder unterschätztes Risiko. Implantologie 22:139–147
Shu C, Li X, Li P (2014) The mechanism of double-stranded DNA sensing through the cGAS-STING pathway. Cytokine Growth Factor Rev 25:641–648. https://doi.org/10.1016/j.cytogfr.2014.06.006
Crapo PM, Gilbert TW, Badylak SF (2011) An overview of tissue and whole organ decellularization processes. Biomaterials 32:3233–3243. https://doi.org/10.1016/j.biomaterials.2011.01.057
Esser E, Brune JC, Pruss A (2016) Peressigsäure-sterilisierte allogene Knochentransplantate zur präimplantatologischen Augmentation des Alveolarfortsatzes. Zeitschrift fur Zahnarztl Implantol:224–232
Breitbach S, Tug S, Simon P (2012) Circulating cell-free DNA: an up-coming molecular marker in exercise physiology. Sports Med 42:565–586. https://doi.org/10.2165/11631380-000000000-00000
Hawthorne AC, Xavier SP, Okamoto R, Salvador SL, Antunes AA, Salata LA (2013) Immunohistochemical, tomographic, and histological study on onlay bone graft remodeling. Part III: allografts. Clin Oral Implants Res 24:1164–1172. https://doi.org/10.1111/j.1600-0501.2012.02528.x
Bauer TW (2007) An overview of the histology of skeletal substitute materials. Arch Pathol Lab Med 131:217–224. https://doi.org/10.1043/1543-2165(2007)131[217:AOOTHO]2.0.CO;2
Hinsenkamp M, Muylle L, Eastlund T, Fehily D, Noël L, Strong DM (2012) Adverse reactions and events related to musculoskeletal allografts: reviewed by the World Health Organisation Project NOTIFY. Int Orthop 36:633–641. https://doi.org/10.1007/s00264-011-1391-7
Ward WG, Gautreaux MD, Lippert DC 2nd, Boles C (2008) HLA sensitization and allograft bone graft incorporation. Clin Orthop Relat Res 466:1837–1848. https://doi.org/10.1007/s11999-008-0294-4
Fretwurst T, Gad LM, Steinberg T, Schmal H, Zeiser R, Amler AK, Nelson K, Altmann B (2018) Detection of major histocompatibility complex molecules in processed allogeneic bone blocks for use in alveolar ridge reconstruction. Oral Surg Oral Med Oral Pathol Oral Radiol pii S2212-4403(18):30054–30053. https://doi.org/10.1016/j.oooo.2018.01.018
Acknowledgments
The authors would like to thank the Erich und Gertrud Roggenbuck foundation, which supported this study through the research grant. Furthermore, the authors thank all patients who participated in this study as well as all the staff who contributed directly or indirectly to the study.
Funding
This study was supported be the Erich und Gertrud Roggenbuck foundation through a research grant no. 0767/114.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards and approved by an ethics committee (Ethik-Kommission der Ärztekammer Hamburg, Hamburg, Germany, PV5211) as well as by the German Registry of Clinical Studies (DRKS 00013010) and were in compliance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Klaus Pantel and Heidi Schwarzenbach should be considered joint senior author.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Solakoglu, Ö., Steinbach, B., Götz, W. et al. Characterization of circulating DNA in plasma of patients after allogeneic bone grafting. Clin Oral Invest 23, 4243–4253 (2019). https://doi.org/10.1007/s00784-019-02867-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00784-019-02867-3