
Abstract
This work is focused on the effects of drying/re-saturation cycles on the structure and components of White Portland Cement (WPC) samples. In particular, WPC of 0.5 water-to-cement ratio was studied by 1H Nuclear Magnetic Resonance (NMR) Relaxometry. A number of NMR sequences and different data processing methods were applied. Short (14 days of re-saturation) and long (6 months for the whole cycle) sorption cycles were investigated, using two drying methods: oven drying and drying under controlled relative humidity. The evolution during drying/re-saturation processes of interlayer space, gel pores, and bigger pores was followed by both the transverse relaxation times and the intensities of the nuclear magnetization. As a result of drying, reversible and irreversible changes in the pore structure, especially concerning the smaller porosity (interlayer and gel pores), were seen. Likewise, 1H signals in the crystalline phases of the cement were investigated by the standard Quadrature Echo method and by a more informative analysis based and a proper acquisition and processing of the longitudinal relaxation data. This analysis allowed the signal separation of 1H nuclei with higher and lower mobility (“solid” components). The NMR signal from this last component was analyzed on the basis of the Pake-Doublet theory in the time domain and two components were clearly detected, and assigned to 1H nuclei of crystalline water in Ettringite and OH groups in Portlandite. Reversible changes of the solid components of the cement samples were observed. This is a new method to deeply investigate the changes of solid components during sorption cycles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Cement-based materials, such as concrete and mortars, are widely used around the world in the building industry. Due to the external environment, these materials are subjected to repeated drying and wetting. Water diffusion through concrete and mortars, and particularly, through the binder phase (cement) largely influences the durability properties of the cement-based materials. The drying of these materials may cause the formation of cracks, which increases the permeability of the material and the attack of acids, sulfates, or alkalis, etc. [1].
The mechanisms involved in the drying process are complex and often interrelated. This is mainly due to the wide range of the pore size distribution in standard cement mixes, which determines, to a large extent, the different transport mechanisms during drying [2, 3]. Thus, studying changes of the porous structure of the cement due to drying/wetting processes could help also in understanding the durability, strength, creep, and shrinkage of the cement-based materials.
Moisture transport within the porous solid involves liquid water and water vapor, subjected to different mechanisms. Examples are the permeation due to a pressure head, the diffusion due to a concentration gradient, the capillary suction due to surface tension acting in the capillaries, or adsorption–desorption phenomena, involving fixation and liberation of molecules on the solid surface. Evaporation and condensation within the porous solid are also important for the determination of the phase in which moisture is transported through the material. All these phenomena may act simultaneously and be predominant in different regions of the cement paste [3]. During saturation cycles, reversible and irreversible changes of the structure were observed and reported in the literature, and it was proposed that irreversible changes mainly occur during the first drying process [4].
The experimental observations most frequently made for sorption studies are based on the adsorption isotherm method: the measurement of the quantity of adsorbed fluid as a function of its vapor pressure at a constant temperature [5]. The “dynamic vapor sorption” shows the sample water content against the relative humidity of the environment (considering the internal relative humidity (RH) of the sample equal to the external). From such experiments, one can determine many important physical properties, such as porosity and its connectivity, specific surface area, transport processes, etc. A drawback for these experiments is that the samples need to undergo preparation by drying, which could greatly alter the material’s original structure [4]. Hysteresis loop in sorption isotherms is a usual phenomenon for cementitious materials even at the low RH level. Feldman and Sereda [5] investigated the hysteresis loop over the RH range (5–100%), and it was clearly irreversible at any point in this RH range and probably even at a lower level.
It has been proposed that hysteresis depends on the pore size distribution of the cement. Schiller et al. [6] stated that for the cement with a smaller number of micropores (< 2 nm), hysteresis is almost not present at low RH. Although, cement with a larger amount of micropores and a small amount of mesopores (2 ÷ 50 nm) have pronounced hysteresis through all the RH range [7]. Following Brouwers, the threshold diameter of gel to capillary pores is about 2.7 nm for cement samples [7]. The hysteresis is also reflected in the values of the volume change of the solid skeleton [6, 7].
Changes in the solid structure of cement samples have been demonstrated as a result of the drying processes. Most of such works showed changes in the cement samples dried at higher than room temperatures [8, 9]. In the work done by Maruyama et al. [10], changes through drying under controlled RH analyzed with MAS NMR technique, have been demonstrated. The component they observed change was assigned to Ettringite, and they proposed that Ettringite could decompose during drying [10].
Although sorption processes of cement materials have been widely studied for many years, there is not a clear understanding of what phenomena are happening inside the cement body. Though many different models for cement sorption exist, there is still no agreement among researchers on a general theory of cement sorption and such phenomena as creep and shrinkage.
Nuclear Magnetic Resonance Relaxometry of 1H nuclei (1H NMR) method is a non-destructive powerful tool that allows the detection of water inside the sample and the monitoring of samples through a long evolution time. NMR allows one to study the pore-space structure (see Sect. 1.1) and solid phases (see Sect. 1.2) of cementitious materials and their evolution during hydration and drying/wetting processes (see Sect. 1.3) [2, 11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26]. Currently, publications related to NMR techniques to study the sorption isotherm process are quite limited and overall, they suffer from the different types of sample preparation.
In this work, the 1H NMR was used to investigate the evolution of cement samples during sorption cycles. It is known that the intensity of the NMR signal is correlated with the amount of water in the pore space as a function of RH [11]. White Portland Cement (WPC) from Aalborg Cement was used with a w/c ratio of 0.5. In particular, as far as we know, the quantitative Pake-Doublet signal analysis in the time domain of the NMR signal, one of the methods we used in this work to characterize the cement changes with RH, is new, not performed previously.
1.1 Characterization of Water in WPC Environments by 1H NMR
NMR Relaxometry methods allow one to study water in different chemical–physical environments inside porous media and in specific cement-based materials, by distinguishing different 1H populations [11, 16, 17, 23,24,25,26,27,28,29]. Using different experimental NMR methods, it is possible to assess the water transport through the pore system, to determine physical properties, such as specific surface area, porosity, and pore size distribution.
The first study of cement by 1H NMR dates back to 1978 when Blinc et al. [12] observed longitudinal and transverse relaxation time (T1 and T2, respectively) evolution through hydration of cement and C3S. In 1985 Schreiner et al. [13] were among the first who separated the NMR signal into two or more components and investigated the hydration process of the cement. Halperin et al. [14] in 1994 applied the NMR fast exchange theory on cement and linked relaxation times with pore sizes and specific surface areas. A lot of different studies were carried out on cementitious materials in the following years [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27], including the study of pore-resolved isotherm by Muller et al. in 2013 [16], the systematic study of the cement pore structure in 2016 by Gajewicz et al. [17], investigations about the evolution of cement paste microstructure during bleeding by Ji et al. [19], and during hydration [23,24,25,26,27], or application of more complex NMR Relaxometry techniques, such as Fast Field Cycling NMR by Korb [18] or 2D-NMR experiments by McDonald et al. [21, 22].
The complex porous structure of the cement paste was characterized by NMR and the T2 relaxation times of 1H components in cement were related to the structural pore sizes [16]. For longer T2 (around 50–100 ms), the water signal was assigned to capillary pores with sizes around 1 µm, the “gel” pores were associated to T2 of 0.3–0.5 ms and pore sizes of 3–5 nm and the interlayer spaces to T2s around 0.1–0.2 ms and size around 1 nm. Solid or crystalline water was characterized by transverse relaxation time in the range of 0.01–0.02 ms [11, 16, 17, 27]. The structure of a hardened WPC paste is shown in Fig. 1 by an SEM image [30] and sketched following the Feldman–Sereda model for the Calcium–Silicate–Hydrate (C–S–H) [5].

For the sorption studies, it was proposed that different mechanisms might play a role at different RH levels, and in pores with different sizes [32]. These mechanisms could be interconnected, which complicates the assessment of the sorption curves. For instance, water in molecular-scale spaces needs to be treated as fundamentally different from that of water in gel and capillary pores. Therefore, it is useful to distinguish different classes of pore sizes and what mechanisms are predominant in each class at different RH levels during sorption cycles.
1.2 NMR Lower (Solid Signal) and Higher Mobility Signal: Pake Analysis of the Solid Signal
In 1948 G.E. Pake showed that it is possible to study a system with rigid pairs of dipolar coupled spin ½ nuclei and to estimate the proton–proton distance from the 1H NMR fine-structure spectrum. By studying gypsum, Pake found that its spectrum forms a typical shape, named “Pake-Doublet” (or “Pake pattern”) [33].
In addition to the polarizing external applied magnetic field B0, neighboring nuclei of a sample influence the magnetic environment of the absorbing nuclei [33] Therefore, the dipolar coupling among 1H nuclei of a sample influences the spectrum of the NMR signal and the corresponding Free Induction Decay (FID). In gases, liquids, and some solids this influence is quite insignificant compared to the inhomogeneities of B0. However, for the crystalline solids, atomic nuclei usually lie in certain lattice positions, and in this case, the interaction of static magnetic dipoles is expected to influence the spectrum line widths and modify the spectrum shape.
Pake showed an analytical expression for such an absorption line, corresponding to a complex FID curve, that, as Abragam has pointed out, bears a strong resemblance to a function which is a combination of a sinc function and a Gaussian broadening term [34]. Look et al. [35] succeeded in computing the Fourier transform of the Pake analytical expression to determine the corresponding function in the time domain G(t) (see Eq. 1). Therefore, in general, the G(t) function could be expressed as the sum of a Gaussian modulated by an oscillating term (the less mobile “solid” component), plus an exponential for the more mobile “liquid” component characterized by the relaxation time \({T}_{2L}^{*}\):
where C(x) and S(x) are the Fresnel integrals, β is the standard deviation of the Gaussian broadening function, which describes the extent of broadening by non-nearest neighbor 1H nuclei, and
where r is the distance between the two interacting nuclei (proton–proton distance or radius), γ is the gyromagnetic ratio, μ0 is the magnetic permeability of free space, and ℏ the Planck constant divided by 2π. The parameter β is the reciprocal of the so-called Gaussian relaxation time (Tg). ML is the amplitude of the “liquid” component and A is a regression parameter proportional to G(0) [35].
The FID shape, as a function of time t, can be, therefore, modeled by Eq. (1).
1.3 Sorption Experiments Studied by NMR
One of the first pore-specific desorption isotherms studied by NMR was performed by Muller et al. [11]. As a result of experiments, the increase of the interlayer spaces was observed during drying and attributed to residual water on the C–S–H surface. From the Quadrature echo (QE) measurements, authors observed an increase in the solid signal, seen at the lower RH level, attributed to the solid-like hydrogen in residual silanol groups. In the research by Maruyama et al. [2], it was found that interlayer signal from NMR experiments increased during drying, reaching a maximum value and decreasing later, while gel and capillary pore signals decreased through the whole drying process. Such changes were attributed to the movable C–S–H sheets that can change spacing and transform from gel pores to interlayer spaces and vice versa. In ref. [17], the explanation of the structural changes was similar and based on “zipping-up” of the interlayer sheets and gel pores collapsing. The redistribution of porosity between finer (< 10 nm) and coarser (> 10 nm) spaces in cement gel after cycles of drying and re-wetting was observed. It was found [17] that coarse pores are created at the expense of collapsing gel pores and that the degree of the changes is linked to the severity of the drying. The reversibility of the structure by re-wetting was observed only after the second sorption cycle [17]. The same conclusion was reached in ref. [28]. It was suggested that [17] “a completely self-consistent picture of all the published data, by us and by others, has not yet emerged”.
2 Materials and Methods
2.1 Materials
For the sorption cycle experiment, WPC from Aalborg Cement was used with the w/c ratio of 0.5. The chemical composition is shown in Table 1 (private communications). Phases less than 0.1% are not included.
The cement paste preparation followed the procedure established elsewhere [36]. The dry cement powder, that was kept at constant low RH in a desiccator, was mixed with distilled water. Then, the paste was cast into a homemade Teflon mold and left to harden for 24 h. After 24 h the sample was taken from the mold and put in the glass tube for the NMR measurements and left for 28 days under-water for hardening. Cement samples hardened for 28 days are in the present manuscript referred to as “as-prepared”.
Some samples were cylindrically shaped with dimensions of circa 1 cm in height and 8 mm in diameter, others were crushed to mm size pieces.
For long sorption processes (~ 6 months for the whole cycle), a homemade RH chamber was used with different chemicals (potassium chloride, silica gel, glycerol) to maintain the RH at specific levels.
Samples, cylindrically shaped or crushed, were progressively dried from 100 to 5% RH and measured at 100, 80, 70, 60, 45, 35, 15, and 5% RH levels. Then, samples were re-saturated in the RH chamber (continuously checked by a data-logger), and measurements were done at 5, 18, 23, 40, 60, 70% RH levels. During re-saturation, a vacuum pump was used to reach 100% RH, and measurements were performed after 1 day and 1 week of re-saturation, respectively. Samples were examined gravimetrically, till mass equilibrium was reached. No specific measures were applied to prevent carbonation. All the experimental procedure for each RH controlled sorption cycle takes up to 6 months.
For the short sorption process (~ 2 weeks of drying), samples were dried in the oven over 60 °C till the mass loss reached a plateau. Samples were re-saturated with the use of a vacuum pump, submerged under-water, and measured up to 14 days of re-saturation.
2.2 NMR Transverse and Longitudinal Relaxation Curves Acquisitions
NMR measurements were done at 25 °C using a relaxometer composed of a permanent magnet (ARTOSCAN, ESAOTE, Genova, Italy) with a magnetic field B0 ≈ 0.2 T (corresponding to 1H Larmor frequency ≈ 8 MHz), a 10 mm solenoid coil, and an NMR console (Stelar s.r.l., Mede, Italy). The 90° pulse width of the spectrometer was 4.5 µs, and the dead time was 25 µs.
T2 relaxation time data were obtained by the use of the Carr–Purcell–Meiboom–Gill (CPMG) sequence with 256–2048 number of echoes, depending on the saturation level of the samples, with an echo time of 60 µs and 200 scans.
T1 relaxation time data were acquired using the logarithmically distributed Aperiodic-Pulse-Sequence Saturation Recovery (LAPSR) [37] pulse sequence. The sequence was performed with the inversion time (TI) increasing logarithmically from 30 µs to 1.6–4 s with 128 blocks (FIDs) and 40–100 scans, depending on the saturation level of the samples.
For the data analysis, to increase the signal-to-noise ratio, instead of a single FID, averages of groups of adjacent FIDs, called stacks, were used. The T1 and T2 quasi-continuous distributions were obtained by the software UpenWin, developed by the Bologna group [38].
2.3 Solid–Liquid Separation, FID Analysis, and Pake-Doublet Analyses
UpenWin software implements an empirical model of the function G(t) to fit each stack of FIDs to separate and quantify the T1 signal of the solid and the liquid parts, where solid corresponds to the Gaussian part of the FID, characterized by the Gaussian time Tg, and liquid corresponds to the exponential part with relaxation time T2FID. It should be noted that in our experiments by “solid” we mean lower mobility protons, such as protons of crystalline water.
The empirical model implemented by UpenWin is given by the following equation:
where Gfrac and Cc are solid-shape FID parameters. Specifically, Gfrac is a coefficient, which corresponds to the Gaussian initial part of the FID, X2 is the corresponding extrapolated amplitude for the solid, X1 is the corresponding extrapolated amplitude for the liquid part. We separated liquid and solid using Eq. 3.
The stacks of FIDs at proper TI times were also examined by the Pake-Doublet analysis using Eq. 1, which represents a more physically founded model. To compute the distance r between the pairs of dipolar interacting 1H nuclei, Eq. 2, was used.
An example of such fitting is shown in Fig. 2 for the WPC sample, dried progressively to 50% RH, where the solid line is the fitted G(t) function by Eq. 1.

Black solid curve is the G(t) function (Eq. 1) fitted to a stack of FIDs acquired from a WPC sample
Using this approach, it was possible to identify which component is responsible for the observed behavior, by comparing the proton–proton distances computed by Eq. 2 with the values obtained from SANS or/and SAXS measurements [23].
For the analysis of the solid component, also a QE sequence was used with echo times from 20 to 36 µs, with a step of 2 µs, 1024 number of points, and 200 scans. The QE protocol experiment and data analysis were developed elsewhere. QE analysis is a robust tool to compare and verify the results obtained by the Pake-Doublet analysis, for example, it is less sensitive to the instrumental deadtime limitation [34].
3 Results and Discussion
3.1 Sorption Cycle from T2 and T1 Experiments
Hysteresis sorption curves were acquired for the WPC samples of w/c = 0.5, for both cylindrical shaped and crushed samples, by both T1 and T2 measurements. An example of the hysteresis sorption curve for the cylindrically shaped sample, dried/wetted at controlled RH, determined by both NMR (T1 signal intensity) and mass measurements, is shown in Fig. 3. The mass value reported in the sorption curve is the mass of the water in the sample. On the right, the plot of the NMR signal intensity vs the mass of water during the drying process is shown.

On the left, sorption curves of the cylindrical WPC sample, dried/wetted at controlled RH, by NMR (total signal intensity from T1 measurements) and by gravimetric analysis (absorbed water mass). On the right, NMR signal intensity is compared to the water content during the drying process
The hysteresis loop is clearly highlighted through the RH range (5–100%) for both, water mass and NMR signal intensity measurements. Although after re-saturation the sample mass returned to its as-prepared value, the NMR signal intensity reached only ≈ 95% of the initial value. This could be attributed to a reduction of the amount of signal detectable by NMR measurements (i.e., part of the water could be no more detectable due to a substantial reduction of its transverse relaxation time, as it might happen for water molecules that turned from liquid to crystalline phase).
A chart to the right of Fig. 3 (NMR signal Intensity vs mass of the water inside the sample) stands for quality control of NMR measurements. From previous works [11], it was concluded that the intensity of the NMR signal is proportional to the amount of water as a function of RH and as we dried the sample and evaporable water is removed, the mass loss should be proportional to the loss of the signal. The linear behavior of the NMR signal-mass relation indicates that all evaporable water was sensed correctly. In addition, as can be seen from Fig. 3, the NMR signal does not start from zero, and this can be ascribed to the interlayer water, detectable by NMR, not lost by the drying process, that remains along with the whole RH range.
It is noteworthy that the same results were obtained for crushed samples, which showed similar trends (data not shown). In addition, sorption curves obtained by T2 measurements (CPMG sequences) showed the same trends (data not shown).
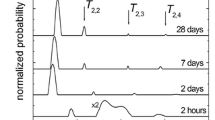
To get a better insight into the evolution of the pore-space structure, it is interesting to follow the behavior of different proton populations through drying and re-saturation separately. With CPMG measurements it is easier to separate the whole signal into different proton populations, as one can run more scans in a relatively short time, significantly improving the signal-to-noise ratio. Figure 4 displays the quasi-continuous T2 distribution obtained by UpenWin for the as-prepared cement sample dried at 100, 80, 70, and 60% RH. Different proton populations, characterized by different T2 classes, can be observed, ascribed to different classes of pores.

T2 relaxation time distributions by UpenWin from CPMG measurements for cement, dried at specific RH points. The intensity of bigger pores is so small that it is not visible in the plot (see Fig. 5)
Figure 5 shows the evolution of the NMR signal intensity, from CPMG measurements, of interlayer space, gel pores, and bigger pores through drying (a) and re-saturation (b) obtained by this kind of analysis on cylindrical samples. Here the signal intensity was normalized from 0 to 1, where 1 is the full signal intensity of the as-prepared sample. The dashed lines are spline interpolations to highlight the trend of the data. The error bar in the graphs corresponds to the 5% estimated by repeated measurements on cement test samples.

Signal intensity normalized to the total signal (from CPMG measurements) of as-prepared sample for different 1H populations (interlayer, gel pores, and bigger pores). Evolution through sorption process at controlled RH, where a drying brunch, b re-saturation
As it is possible to see from Fig. 5a, the signal assigned to the gel pores water decreased through all the drying steps. Through the re-saturation process (Fig. 5b), it is seen that the gel pores water signal increased through all the steps. However, even waiting for a 1-week re-saturation under-water, the signal intensity of the gel pores was less than for the as-prepared sample. It seems, therefore, that gel pores have undergone irreversible changes.
The signal assigned to the interlayer water, after an increase, seems to decrease when RH < 30%. Due to the signal scattering and approximations of the data processing, more investigations will be needed to corroborate this behavior. The results of the full re-saturation process are shown in Fig. 5b. At the end of the re-saturation process under-water the whole CPMG signal of the cement sample re-established a value comparable with the value of 95% obtained by LAPSR and it is consistent with the data shown in Fig. 3. Nevertheless, this result resembles the observation reported in ref. [2]. Moreover, the analysis of the T2 values (Fig. 6) strengths the data in Fig. 5a. As observed for the signal intensity, also the T2 of interlayer water started to decrease from the initial value of 180 µs (as-prepared value) for the fully saturated sample to 90 µs for 15% RH drying condition (Fig. 6a). The decrease of T2 is consistent with the shrinking of the interlayer space. The NMR signal assigned to the bigger pores was negligible below 80% RH.

Evolution of the T2 relaxation time of interlayer water through a drying, b re-saturation
Through all the re-saturation points, the T2 relaxation time of the interlayer space slowly increased as it is shown in Fig. 6, but even after 1 week of re-saturation under-water the T2 was shorter (≈150 µs) than in the as-prepared state (see Fig. 6b), as well as the signal intensity.
The behavior of the interlayer spaces during drying and re-saturation may suggest the collapse of the C–S–H sheets during drying. As suggested by the T2 values (see Fig. 6), it seems that some of the interlayer spaces could not re-establish back or need some longer time of re-saturation than in these experiments. Such irreversible change of the interlayer spaces is in good agreement with results from the literature [16].
In summary, the behavior of the interlayer spaces and gel pores suggests irreversible changes in the cement structure.
3.2 Sorption Cycle with Analysis of Solid Components (Pake-Doublet and QE)
The behavior of solid components was examined using both Pake-Doublet analysis and QE.
An example of the UpenWin analysis of the solid component, separated from the liquid as described in paragraph 2.3, is presented in Fig. 7 for a cylindrical sample, where the T1 distributions of the solid component are shown for 100% and 15% RH levels.

T1 distribution of the solid component computed by UpenWin after solid–liquid separation
The solid signal appeared to be due to two different components: one with shorter T1 (solid-short, labeled as SS) of the order of a few milliseconds or less and another one (solid-long, labeled as SL) with longer T1 of the order of hundreds of milliseconds.
These solid components were obtained and discussed for the first time in ref [23]. The SL population was ascribed to rigid pairs of 1H nuclei belonging to two classes of molecules whose 1H nuclei have a very low chance to interact with liquid water, having the typical relaxation times of “solids” (T2 of the order of tens of μs and T1 of the order of seconds), and precisely to 1H in Portlandite and in the crystalline water of Ettringite. The SS population appeared to have 1H more mobile than in SL, as revealed by the pure Gaussian behavior and was ascribed to Ca–OH and/or anisotropically oriented water molecules inside the layers of C–S–H gel [23].
Stacks of FIDs, which have a clear Pake-Doublet pattern behavior (see Fig. 2) correspond to inversion times TI in the LAPSR sequence no shorter than 5 ms, meaning that these FIDs are contributed only by the SL component; therefore, only the SL component will be discussed.
The SL component obtained from the stacks with T1 ≈ 200 ms was analyzed by Eq. 1. A significant improvement of the fit was obtained by assuming that the component SL was composed of two Gaussian populations with two different Tg values (TgE and TgP), and two different corresponding proton–proton distances, rE and rP calculated using Eq. 2. The results obtained for the WPC samples, dried at controlled RH, are shown in Table 2.
Essentially, Gaussian times and distances did not substantially change through drying, for both populations. Comparing these values of proton–proton distances with previously done work [23], the first component TgE with distance rE around 1.5 Å was assigned to crystalline water in Ettringite, and the second component TgP with distance rP around 1.9 Å was assigned to 1H of (OH)2 groups in Portlandite. Figure 8 shows the evolution of the signal intensity (in percent respect to the total solid signal) of the two components TgE and TgP.

Evolution of the signal (in percent) of the two components belonging to the SL signal, characterized by the two Gaussian times TgE (Ettringite) and TgP (Portlandite), through drying at controlled RH. Dashed lines are an interpolation
From Fig. 8, it seems that the amount of the Ettringite decreased up to about 40% RH and then later increased, which is in good agreement with results obtained by MAS technique by Maruyama et al. [10]. On the contrary, the component, characterized by TgP (and rP), and assigned to Portlandite, did not seem to change significantly through drying.
Samples, dried in the oven or dried at controlled RH, showed similar behavior with the same values for Tg relaxation time and proton–proton distance. For these samples, the fraction of the solid signal related to the TgE component decreased up to several days of drying and then later it increased.
After around 2 months of drying in the oven, samples were fully re-saturated under-water with the use of a vacuum pump method. After 2 weeks of re-saturation, the solid signal associated with Ettringite did not differ significantly from the as-prepared value, as shown in Fig. 9.

Evolution of the NMR signal due to crystalline water 1H in Ettringite through drying/re-saturation cycle
It could be of interest to point that, by comparing these results with those obtained by CPMG measurements, the Ettringite signal intensity seems to increase around at the same drying condition (< 40% RH), at which the T2 of the interlayer water starts to decrease (see Fig. 6).
For comparison, the analysis of the solid component was performed also by QE. In Fig. 10 the evolution of the solid signal [normalized intensity respect the total QE signal (a), and TgQE (b)] through drying at controlled RH obtained by the QE measurements is shown.

Evolution of the solid signal of cement through drying at controlled RH from QE measurements, a normalized signal intensity, b TgQE relaxation time
From Fig. 10b, it is seen that the TgQE relaxation time increased through drying. This is in apparent contrast with the results shown in Fig. 8. It is worth noting that by the QE method is not possible to distinguish two SL populations, as well as to separate SL and SS components. The observed effect might be due to the increase of the SS component, better detectable, because the sample was drying, and therefore, the solid signal was not “covered” by the liquid part of the signal. In addition, for the behavior of TgQE in Fig. 10b, one has to take into account that, as observed before, with the QE procedure it was not possible to separate the signal into SL and SS components and what it is shown is their average Gaussian time. The apparent contrast is resolved if one considers that Fig. 10a shows the signal amplitude of the whole solid phase, while Fig. 8 shows the behavior of two separate components.
4 Conclusion
In this work the effects of drying/re-saturation cycles on the structure and components of White Portland Cement samples were studied by NMR Relaxometry of 1H nuclei, with particular focus on the evolution during drying/re-saturation processes of three cement pore classes (interlayer, gel pores, and bigger pores) and of the solid components Ettringite and Portlandite. Our goal was to increase the current understanding of the changes occurring during drying and the first re-wetting cycle, by integrating the information about the redistribution of water among components in two ways: (1) to study the evolution of these classes of porosity by both signal NMR intensity and T2 relaxation time distributions; (2) to study the evolution of the signal from the crystalline phases Ettringite and Portlandite. The results are listed as follows.
-
I.
Results about the evolution of the porous structure.
The signal of 1H nuclei of water in the pore space was analyzed by both T1 and T2 measurements.
-
1.
Hysteresis sorption curves obtained for the WPC samples by both T1 and T2 measurements are in agreement with literature.
-
2.
The evolution of interlayer space, gel pores, and bigger pores through drying and re-saturation obtained by this CPMG experiments on the WPC samples during drying at controlled RH and in the oven are in good agreement with what reported in the literature [16].
-
3.
We analyzed CPMG decay data with quasi-continuous T2 distribution, instead of multi-exponential four-component analysis with fixed T2 values [16]. Our choice allowed us to observe that the T2 values of the different pore classes changed during re-wetting instead of remaining constant.
-
4.
We were able to observe the decrease of the T2 of the interlayer value during drying, from 180 µs to about 90 µs, a clear fingerprint of the progressive shrinking of these spaces, and, more interesting, that it remained constant at about 90 µs during the rewetting process, and never recovered the as-prepared value. This is a clear visualization of the irreversible shrinkage of the interlayer spaces occurring during the first sorption cycle.
In summary, some irreversible changes of the pore space are happening during the first sorption cycle, largely contributed by the changes of interlayer spaces and gel pores. The quasi-continuous relaxation time analysis allowed us to get a deeper insight on the irreversible shrinkage of the interlayer spaces. The same behavior was observed for intact and crushed samples.
-
II.
Results about the evolution of the solid components.
-
1.
The signal of 1H nuclei in the crystalline phases of the cement was investigated by the standard Quadrature Echo method and by a more informative analysis based and a proper acquisition and processing of the longitudinal relaxation data. This methodology was based on measurements of the T1 relaxation by the sequence LAPSR. By this sequence FID signals at different inversion times can be acquired. By means of a proper analysis of the FIDs, the solid–liquid separation method can be applied. Then, a further analysis of the solid signal can be performed to discriminate different solid (lower mobility) 1H components. The most important results can be listed as follows. The signal from the lower mobility component was analyzed by the Pake-Doublet theory and two components were clearly detected, assigned to 1H nuclei of crystalline water in Ettringite and OH groups in Portlandite.
-
2.
The crystal water in Ettringite, interpreted with Pake-Doublet analysis, showed a similar trend during drying at relative humidity and in the oven. These behavior of water in Ettringite is similar to what was found by Maruyama et al. [10], using different methods.
In conclusion, 1H NMR Relaxometry methods are a good tool to be used for the further exploration of pore-space and solid component changes in cementitious materials, induced by sorption cycles. In particular, it has been shown that these methods not only allow one to track the behavior of the solid components but also to identify different solid phases. As far as we know, quantitative analyses of Pake-Doublets signals in cement sorption cycle experiments and the possibility to distinguish Ettringite from Portlandite NMR signals during these processes were not performed previously. Further experiments are in progress to corroborate these results, especially those concerning the changes observed around specific RH values.
Availability of Data and Materials
The data that support the findings of this study are available from the corresponding author upon request.
Code Availability
The software UpenWin is marketed by the University of Bologna.
References
V.G. Papadakis, C.G. Vayenas, M.N. Fardis, Physical and chemical characteristics affecting the durability of concrete. Aci Mater. J. 88(2), 186–196 (1991)
I. Maruyama, T. Ohkubo, T. Haji, R. Kurihara, Dynamic microstructural evolution of hardened cement paste during first drying monitored by 1H NMR relaxometry. Cem. Concr. Res. 122, 107–117 (2019). https://doi.org/10.1016/j.cemconres.2019.04.017
A.E. Idiart, Coupled analysis of degradation processes in concrete specimens at the meso-level. Doctoral Thesis, Doctoral Program in Geotechnical Engineering Barcelona, 2009
V. Baroghel-Bouny, Water vapour sorption experiments on hardened cementitious materials: part I: essential tool for analysis of hygral behaviour and its relation to pore structure. Cem. Concr. Res. 37(3), 414–437 (2007). https://doi.org/10.1016/j.cemconres.2006.11.019
R.F. Feldman, P.J. Sereda, A model for hydrated Portland cement paste as deduced from sorption-length change and mechanical properties. Matériaux et Constr. 1, 509–520 (1968). https://doi.org/10.1007/BF02473639
P. Schiller, M. Wahab, Th. Bier, S. Waida, H.J. Mögel, Capillary forces and sorption hysteresis of cement pastes with small slit pores. Procedia Mater. Sci. 11, 649–654 (2015). https://doi.org/10.1016/j.mspro.2015.11.010
H.J.H. Brouwers, The work of powers and Brownyard revisited: Part 1. Cem. Concr. Res. 34(9), 1697–1716 (2004). https://doi.org/10.1016/j.cemconres.2004.05.031
Y. Aono, F. Matsushita, S. Shibata, Y. Hama, Nano-structural changes of C-S-H in hardened cement paste during drying at 50˚C. J. Adv. Concr. Technol. 5(3), 313–323 (2007). https://doi.org/10.3151/jact.5.313
A. Bentur, R.L. Berger, F.V. Lawrence, N.B. Milestone Jr., S. Mindess, J.F. Young, Creep and drying shrinkage of calcium silicate pastes III. A hypothesis of irreversible strains. Cem. Concr. Res. 9(1), 83–95 (1979). https://doi.org/10.1016/0008-8846(79)90098-X
I. Maruyama, Y. Nishioka, G. Igarashi, K. Matsuic, Microstructural and bulk property changes in hardened cement paste during the first drying process. Cem. Concr. Res. 58, 20–34 (2014). https://doi.org/10.1016/j.cemconres.2014.01.007
A.C.A. Muller, K.L. Scrivener, A.M. Gajewicz, P.J. Mcdonald, Use of bench-top NMR to measure the density, composition and desorption isotherm of C-S–H in cement paste. Microporous Mesoporous Mater. 178(15), 99–103 (2013)
R. Blinc, M. Burgar, G. Lahajnar, M. Rozmarin, V. Rutar, I. Kocuvan, J. Ursic, NMR relaxation study of adsorbed water in cement and C3S pastes. J. Am. Ceram. Soc. 61(1–2), 35–37 (1978)
L.J. Schreiner, J.C. Mactavish, L. Miljkovic, M.M. Pintar, R. Blinc, G. Lahajnar, D.D. Lasic, L.W. Reeves, NMR line shape-spin-lattice relaxation correlation study of Portland cement hydration. J. Am. Ceram. Soc. 68(1), 10–16 (1985)
W.P. Halperin, J.-Y. Jehng, Y.-Q. Song, Application of spin-spin relaxation to measurement of surface area and pore size distributions in a hydrating cement paste. Magn. Reson. Imaging 12(2), 169–173 (1994)
L. Pel, K. Hazrati, K. Kopinga, J. Marchand, Water absorption in mortar determined by NMR. Magn. Reson. Imaging 16(5–6), 525–528 (1998)
A.C.A. Muller, K.L. Scrivener, A.M. Gajewicz, P.J. McDonald, Densification of C-S–H measured by 1H NMR relaxometry. J. Phys. Chem. 117, 403–412 (2013)
A.M. Gajewicz, E. Gartner, K. Kang, P.J. McDonald, V. Yermakou, A 1H NMR relaxometry investigation of gel-pore drying shrinkage in cement pastes. Cem. Concr. Res. 86, 12–19 (2016). https://doi.org/10.1016/j.cemconres.2016.04.013
J.-P. Korb, NMR and nuclear spin relaxation of cement and concrete materials. Curr. Opin. Colloid Interface Sci. 14(3), 192–202 (2009)
Y. Ji, L. Pel, Z. Sun, The microstructure development during bleeding of cement paste: an NMR study. Cem. Concr. Res. 125, 105866 (2019). https://doi.org/10.1016/j.cemconres.2019.105866
J.P. Korb, Multiscale nuclear magnetic relaxation dispersion of complex liquids in bulk and confinement. Prog. Nuclear Magn. Reson. Spectrosc. 104, 12–55 (2018). https://doi.org/10.1016/j.pnmrs.2017.11.001
P.J. McDonald, J.-P. Korb, J. Mitchell, L. Monteilhet, Surface relaxation and chemical exchange in hydrating cement pastes: a two-dimensional NMR relaxation study. Phys. Rev. E 72, 011409 (2005). https://doi.org/10.1103/PhysRevE.72.011409
L. Monteilhet, J.P. Korb, J. Mitchell, P.J. McDonald, Observation of exchange of micropore water in cement pastes by two-dimensional nuclear magnetic resonance relaxometry. Phys. Rev. E, Am. Phys. Soc. 2006
M. Gombia, V. Bortolotti, B. De Carlo, R. Mongiorgi, S. Zanna, P. Fantazzini, Nanopore structure buildup during endodontic cement hydration studied by time-domain nuclear magnetic resonance of lower and higher mobility 1H. J. Phys. Chem. B 114, 1767–1774 (2010). https://doi.org/10.1021/jp907248r
V. Bortolotti, P. Fantazzini, R. Mongiorgi, S. Sauro, S. Zanna, Hydration kinetics of cements by time-domain nuclear magnetic resonance: application to Portland-cement-derived endodontic pastes. Cem. Concr. Res. 42, 577–582 (2012). https://doi.org/10.1016/j.cemconres.2011.12.006
V. Bortolotti, R.J.S. Brown, P. Fantazzini, M. Mariani, Evolution of a short-T2 liquid-like 1H signal during the hydration of white Portland cement. Microporous Mesoporous Mater. 178, 108–112 (2013). https://doi.org/10.1016/j.micromeso.2013.03.010
V. Bortolotti, L. Brizi, R.J.S. Brown, P. Fantazzini, M. Mariani, Nano and Sub-nano multiscale porosity formation and other features revealed by 1H NMR relaxometry during cement hydration. Langmuir 30, 10871–10877 (2014). https://doi.org/10.1021/la501677k
P.J. McDonald, O. Istok, M. Janota, A.M. Gajewicz-Jaromin, D.A. Faux, Sorption, anomalous water transport and dynamic porosity in cement paste: a spatially localised 1H NMR relaxation study and a proposed mechanism. Cem. Concr. Res. 133, 106045 (2020). https://doi.org/10.1016/j.cemconres.2020.106045
N.N. Fischer, R. Haerdtl, P.J. McDonald, Observation of the redistribution of nanoscale water filled porosity in cement based materials during wetting. Cem. Concr. Res. 68, 148–155 (2015)
A. Zhang, W. Yang, Y. Ge, Y. Wang, P. Liu, Study on the hydration and moisture transport of white cement containing nanomaterials by using low field nuclear magnetic resonance. Constr. Build. Mater. (2020). https://doi.org/10.1016/j.conbuildmat.2020.118788
N. Shiferaw, L. Habte, T. Thenepalli, J.W. Ahn, Effect of eggshell powder on the hydration of cement paste. Materials 12, 2483 (2019). https://doi.org/10.3390/ma12152483
J.H.M. Jennings, Refinements to colloid model of C-S-H in cement: CM-II. Cem. Concr. Res. 38(3), 275–289 (2008)
M.B. Pinson, E. Masoero, P.A. Bonnaud, H. Manzano, Q. Ji, S. Yip, J.J. Thomas, M.Z. Bazant, K.J. Van Vliet, H.M. Jennings, Hysteresis from multiscale porosity: modeling water sorption and shrinkage in cement paste. Phys. Rev. Appl. (2015). https://doi.org/10.1103/PhysRevApplied.3.064009
G.E. Pake, E.M. Purcell, Lineshapes in nuclear paramagnetism, Phys. Rev. 74 (1948)
A. Abragam, The Principles of Nuclear Magnetism (Clarendon Press, 1983), p. 618
D.C. Look, I.J. Lowe, J.A. Northby, Nuclear magnetic study of molecular motions in solid hydrogen sulfide. J. Chem. Phys. 44, 9 (1966)
P.J. McDonald, A.M. Gajewicz, Good Practice Guide No. 144, The characterisation of cement based materials using T2 1H nuclear magnetic resonance relaxation analysis, University of Surrey, 2016, pp. 62–65
S. Sykora, E. Vacchelli, V. Bortolotti, P. Fantazzini, New Universal NMR Sequences: PERFIDI and LAPSR, GIDRM—XXXVI National Congress on Magnetic Resonance, Salerno (Italy), 2006. https://doi.org/10.3247/SL1Nmr06.006
V. Bortolotti, R.J.S. Brown, P. Fantazzini, UpenWin: a software to invert multi-exponential decay data, http://www.unibo.it/PortaleEn/Research/Services+for+companies/UpenWin.htm
Acknowledgements
Work supported by Horizon 2020—Marie Skłodowska-Curie—Innovative Training Network (ITN)—2017 Actions. Grant Agreement no. 764691.
Funding
Open access funding provided by Alma Mater Studiorum - Università di Bologna within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagmutdinova, A., Brizi, L., Fantazzini, P. et al. Investigation of the First Sorption Cycle of White Portland Cement by 1H NMR. Appl Magn Reson 52, 1767–1785 (2021). https://doi.org/10.1007/s00723-021-01436-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-021-01436-w