
Abstract
High-resolution two-dimensional (2D) 1H–13C heteronuclear correlation spectra are recorded for selective observation of interfacial 3–5.5 Å contacts of the uniformly 13C-labeled phycocyanobilin (PCB) chromophore with its unlabeled binding pocket. The experiment is based on a medium- and long-distance heteronuclear correlation (MELODI–HETCOR) method. For improving 1H spectral resolution, a windowed phase-modulated Lee–Goldburg (wPMLG) decoupling scheme is applied during the t 1 evolution period. Our approach allows for identification of chromophore–protein interactions, in particular for elucidation of the hydrogen-bonding networks and charge distributions within the chromophore-binding pocket. The resulting pulse sequence is tested on the cyanobacterial (Cph1) phytochrome sensory module (residues 1–514, Cph1Δ2) containing uniformly 13C- and 15N-labeled PCB chromophore (u-[13C,15N]-PCB-Cph1Δ2) at 17.6 T.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
High-resolution solid-state nuclear magnetic resonance (SSNMR) has become a powerful spectroscopic tool for the characterization of structure and dynamics in non-crystalline or low-soluble protein complexes [1–3]. This technique has been further developed to obtain chemical shift assignments [4–6] and to probe distance restraints in selectively isotope-labeled constituents of proteins [7–9]. However, most of these studies focused on well-resolved homo- and heteronuclear correlation (HOMCOR and HETCOR) spectra involving 13C and 15N nuclei. As the most naturally abundant NMR observable nuclear spin, 1H NMR has not yet been exploited sufficiently to characterize proteins in the solid state, which suffers from low spectral resolution due to the severe homogeneous broadening (up to 50 kHz) [10] involving the strong 1H–1H dipolar couplings. To remove the homonuclear dipolar interactions among protons, various combinations of multiple radio-frequency (RF) pulses have been proposed, typically under the condition of magic-angle spinning (MAS) [11], known as 1H CRAMPS (combined rotation and multiple pulse spectroscopy). At the moderate spinning frequency (10–15 kHz), Lee–Goldburg (LG) based homonuclear decoupling schemes, such as frequency-switched LG (FSLG) [12, 13] and phase-modulated LG (PMLG) [14, 15], are the most commonly used CRAMPS methods today for 1H observation in both one-dimensional (1D) and 2D fashions. Recently, acquisition windows have been inserted in PMLGn and DUMBO (decoupling using mind-boggling optimization) classes of sequences, leading to the corresponding wPMLGn [16–18] and wDUMBO-1 [19] series, which have been proven efficient for a variety of applications [10, 20, 21]. On the other hand, the information available from 1H SSNMR is of great value for investigation of intermolecular interactions, such as strong/weak hydrogen-bonding [22–24] and π–π packing arrangements [25].
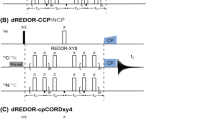
Functionally crucial interactions between proteins and cofactors in their binding pockets may be explored in some detail by 2D 1H–13C heteronuclear correlation NMR spectroscopy. The efficiency of such HETCOR experiments depends primarily on the line width obtained in 1H indirect dimension. Here, we report a 1H sensitivity-enhanced high-resolution HETCOR experiment (Fig. 1a) for recognizing the intermolecular contacts between a uniformly 13C-labeled chromophore and its protein matrix at the natural abundance through the detection of medium- and long-range interfacial connectivities between 1H and 13C spins. Our method was built on the basis of a MELODI–HETCOR experiment [26, 27]. The sensitivity enhancement of 1H dimension was achieved through the use of windowed PMLG homonuclear decoupling over t 1 evolution, in our case, wPMLG3+ (Fig. 1b).

a Pulse sequence used for long-range 1H–13C interfacial correlation experiment with wPMLG3+ 1H decoupling scheme. b Schematic representation of 1H{wPMLG3+} decoupling irradiation during t 1. For each t 1 increment, an additional wPMLG3+ block is added. See the text for more details
The MELODI–HETCOR experiment considerably simplifies the conventional 1H–13C HETCOR spectra [28] by filtering out the mostly trivial one-bond 1H–13C correlations with rotational echo double-resonance (REDOR) dephasing technique [29] while retaining the long-range, structurally revealing heteronuclear correlations. In this experiment, 1H magnetization is transferred to 13C spins by Lee–Goldburg cross-polarization (LG-CP), which can be used to determine short-range 1H–13C heteronuclear distances precisely (i.e., within the limit of 3 Å) from the coherent transfer occurring during short LG-CP contact times [30]. To probe the long-range intermolecular correlations, on the other hand, one requires a relatively long transfer time due to the low heteronuclear dipolar coupling of ~150 Hz [31, 32]. This method is particularly appropriate for exploring fractionally labeled protein (e.g., the uniformly 13C-labeled chromophore interacting with the other completely unlabeled components as performed in this paper), whereas for 1H homonuclear decoupling strategy, a windowless LG-based acquisition method (FSLG) was applied in the MELODI experiment [26]. There is, thus, plenty of room for improvement [20, 33].
2 Materials and Methods
2.1 Sample Preparation
Preparation of uniformly 13C- and 15N-labeled PCB chromophore, Cph1Δ2 apo- and holoprotein was described elsewhere [34–36]. For NMR measurements of Pr Cph1Δ2 phytochrome at 100% occupancy, the sample was pre-irradiated with light from an array of far-red light-emitting diodes (LEDs) (λ max ≈ 730 nm, 20 nm full-width at half-maximum (FWHM)) surrounding a 1-mm bore glass capillary syringe.
2.2 Pulse Sequence
The pulse sequence used to record high-resolution 1H–13C interfacial correlation NMR spectra is shown in Fig. 1a. The experiment starts with a REDOR block, lasting for two rotor cycles (~169 μs in our case), during which 13C-attached 1H spins are dephased. By the end of the constant dipolar dephasing period, a 1H35°−y pulse positions the cosine-modulated magnetization in the plane perpendicular to the effective RF field. A subsequent 90° 1H excitation pulse prior to t 1 evolution (“effective z-rotation”) is employed for the proper off-resonance regime [17, 18]. Then, the wPMLG3+ CRAMPS homonuclear decoupling scheme is introduced for 1H evolution and, at end of the t 1 encoding period, another magic-angle pulse, 35°+y, is applied to bring the z-magnetization to the effective field of the spin-lock pulse for polarization transfer to 13C. The following ramped amplitude LG-CP segment [39] is kept relatively long (e.g., 2.3 ms) to survive the weak transfer processes of 1H magnetization over long distances, and 13C magnetization evolves under 1H TPPM decoupling [40] as in the conventional HETCOR experiments.
The wPMLG3+ scheme for homonuclear decoupling during t 1 evolution is depicted in Fig. 1b. In wPMLG3 decoupling, the basic unit consists of PMLG3 block (τ PMLG3) and the acquisition window τ w (as shown in Fig. 1b by an asterisk), thus the decoupling cycle period is τ c = τ PMLG3 + τ w. The PMLG3 block is composed of two LG units (τ LG), each one divided into three sequential pulses with the RF phase (φ i ) increment of 69.27° (Δφ = 207.8/n, with n = 3) and starting with 34.63° (Δφ/2) [17]. For the phase symmetrization argument, the RF phases in the second LG unit are switched by 180° (e.g., φ 3′ = φ 3 + 180°). Hence, PMLG3+ block is accomplished in a six-step phase cycle of 34.63°, 103.90°, 173.17°, 353.17°, 283.90° and 214.63°. The wPMLG3+ decoupling efficiency correlates to several experimental parameters, like RF field power (ν 1), unit pulse length (Δt), acquisition windows (τ w), rotation frequency (ν r), and 1H RF offset frequency (Δν). The optimizations were carried out by acquiring 1D 1H wPMLG3+ MAS spectra on glycine through monitoring the splitting between α-protons lines [18]. To optimize performance for 2D fashion, ν 1 and Δt, were further examined in the indirect dimension of 2D 1H{wPMLG3+}–1H{wPMLG3+} homonuclear correlation spectra of the same sample [33]. Optimum values obtained using a pulse length Δt of 2.1 μs and RF amplitude (ν 1) of 78 kHz. In addition, the acquisition window τ w was set to 6.5 μs, slightly higher than the calibrated value, for compensation of the spectral quality loss caused by any mistuned wobble curve [33]. The PMLG3 segment was composed of six RF pulses (Δt = 2.1 μs) with a delay τ x of 0.1 μs [18] between two pulses to stabilize their phases, thus τ PMLG3 = 13.2 μs. The overall length of decoupling cycle time (τ c) of wPMLG3+ pulse unit was equal to 19.7 μs (τ c = τ PMLG3 + τ w), thus the characteristic frequency (ν c) of the wPMLG3+ CRAMPS cycle was 50.76 kHz. To avoid an undesirable destructive interference between MAS and spin space modulation, the rotor period (τ r) have been chosen to meet the condition of 3.5 < τ r/τ c < 5, and τ r ≠ 4τ c [16, 37, 41]. For RF power levels available in our AV-750 WB spectrometer using 4-mm triple-resonance MAS probe, 10–13.5 kHz spinning speed is commonly used. The rotation frequency (ν r) of 11.778 kHz (τ r = 84.90 μs, thus τ r/τ c ≈ 4.31) was found to be optimal for the spectral resolution with 1H RF field strength (ν 1) of 78 kHz, and to be sufficiently distant from obvious degeneracy number, τ r/τ c = 4. Moreover, 1H RF offset frequency (Δν) of −6,000 Hz was set to keep spiky resonance lines (e.g., artifacts from rotor and RF lines) from falling over the spectrum [18].
2.3 MAS NMR Experiment
The 2D 1H–13C interfacial correlation spectrum shown in Fig. 3 was obtained from u-[13C,15N]-PCB-Cph1Δ2 in the Pr state at 233 K on a Bruker AV-750 WB spectrometer (Karlsruhe, Germany), equipped with 4-mm triple-resonance CP/MAS probe (Bruker). Approximately 15 mg (≈80 μL) of protein was packed into a 4-mm zirconia MAS rotor. There is no significant loss of the 1H spectral resolution when compared with the sample restricted to the central part of the rotor (that is, CRAMPS rotor with the sample volume of ~12 μL), but the Gaussian line shape is slightly disturbed [37]. The spinning frequency was 11,778 ± 3 Hz, regulated by a pneumatic control unit. Optimized 1H 90° and 13C 180° pulse lengths were obtained at 3.1 and 5.2 μs, respectively. The dipolar filter period of 38 μs was set to effectively suppress the 1H–1H homonuclear dipolar interaction [26]. The spectrum was acquired with a 2.3-ms LG-CP contact time, which is sufficiently long to detect the interfacial 1H correlations of the chromophore up to ~4.5 Å [31]. However, the cut-off distance used to define the potential 1H–13C correlations was set to 5.5 Å due to the thermal motion of protein atoms on the timescale of picoseconds, expressed as root-mean-square (rms) fluctuations (≈0.5 Å in average for each atom) [38]. The 1H power was ramped 80–100% during CP, and 1H RF field strength for TPPM decoupling was about 80 kHz. The spectrum was recorded with 3,702 transients with a relaxation delay of 1.536 s for each t 1 slice, and a total of 120 slices combined in the indirection dimension, leading to a total experimental time of ca. 138 h. The t 1 data was recorded in an off-resonance manner and linear predicted by 32 points using 40 LP coefficients. A 90° shifted squared sine bell window function was applied, and zero-filled to 2,048 points prior to Fourier transformation. The t 2 data was zero-filled to 4,096 points and 50 Hz line broadening for exponential multiplication. The data were processed with Bruker Topspin 2.1 and further analyzed by using the Sparky 3.114 (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, CA). Carbon resonance frequency (ω 1) and proton resonance frequency (ω 2) are in parts per million (ppm).
3 Results and Discussion
The performance of the resulting pulse sequence is demonstrated on u-[13C,15N]-PCB-Cph1Δ2 (~58 kDa) in the Pr state. In all phytochromes, an open-chain tetrapyrrole chromophore (Fig. 2b) is covalently attached to a cysteine residue of the apoprotein [42, 43]. Irradiation causes the chromophore to undergo a photoconversion between the ground state Pr (red-light absorbing, λ max ~ 658 nm in the case of Cph1) and the photoactivated far-red-absorbing state Pfr (λ max ~ 702 nm for Cph1) via a series of intermediates [44–46]. Various spectroscopic studies have demonstrated that the Pr → Pfr photoactivation begins with a Z → E isomerization of the methine bridge between rings C and D [6, 47–50] or, possibly, rings A and B [51] (Fig. 2a). The discrepancy in assigning the position of photoisomerization perhaps arises from the different phytochromes being studied (canonical PAS–GAF–PHY photosensor from temperate species vs. a “naked” GAF domain from a thermotolerant species in Ref. [51]). It has been demonstrated that the phototransformation is not a local event, but dramatically modifies the chromophore–protein interaction in a wide range [6]. Thus, it is of importance to study chromophore–protein interactions to clarify the role of the chromophore-binding pocket during the photocycle and the mechanism underlying the subsequent intramolecular signal transduction pathway.

a Structural formula of protein-bound PCB shown in the ZZZssa configuration. The tetrapyrrole rings and representative PCB atoms are labeled for reference. b PCB-binding pocket in Cph1Δ2 [52]. Structural elements of the protein domains involved in chromophore binding (GAF domain, tongue region from PHY domain and N-terminal α1-helix from PAS domain) are indicated
Characteristic datasets for u-[13C,15N]-PCB-Cph1Δ2 in the Pr state are shown in Fig. 3. The 13C chemical shifts are well in line with the values reported formerly by our group (Δσ C ≤ 0.2 ppm) [6]. In the region of 160–190 ppm (Fig. 3a), four 13C chromophore resonances are detected, locating at 184.1 ppm (C1 of ring A, for numbering, see Fig. 2a), 180.1 ppm (C83, ring B), 179.0 ppm (C123, ring C) and 172.7 ppm (C19, ring D), respectively. The structure of this protein recently resolved at a 2.45-Å resolution (PDB code 2VEA) [52] provides a reliable template for assigning the long-range Pr heteronuclear correlation signals. For each chromophore carbon atom, we measured all possible intermolecular residue contacts within the enclosure sphere up to ~5.5 Å. As an example given, at least eight 1H contacts can be distinguished for the D-ring carbonyl (C19, Fig. 3a) indicating the strong interactions with amino acids in its proximity. More specifically, Cph1 2VEA Pr structure shows that ring D forms a strong hydrogen bond via this carbonyl group with the imidazole moiety of the highly conserved His-290. The build-up kinetics of a long-range intermolecular correlation primarily depends on the heteronuclear distance [26, 27, 30]. Thus, the cross-peak intensities can provide a fair estimation of the 1H–13C proximities. Therefore, we assign this interaction (His-290 HNε2···C19) to the most intense cross-peak centered at 12.7 ppm (labeled as ‘H290 Nε2’) in the downfield 1H frequencies of 10–13 ppm, which is confirmed by 1H correlations with the adjacent carbons of the C19 (indicated by the horizontal dashed line C), e.g., His-290 HNε2···C18 (Fig. 3b) having a crystal distance of 4.60 Å, and with the ethyl side-chain, C181 (5.10 Å) and C182 (4.29 Å) of the ring D (Fig. 3d), locating at 134.2, 16.5 and 13.2 ppm in the ω 2-dimension, respectively. In contrast, the correlation signal involving His-290 HNε2···C17 (Fig. 3b) is not completely resolved, since they are 5.47 Å apart in 2VEA, close to the limit of the transfer range. Another perfectly conserved histidine residue locating above the chromophore, His-260, exhibits many correlations with the chromophore. The 1H correlations at 10–18 ppm in the C10 slice (Fig. 3c), for instance, identify the protonation shift between positively charged and neutral imidazole of His-260 in the Pr state. The 1H chemical shifts of HCδ2 (‘H260Cδ2-I’) and HNε2 (‘H260 Nε2-I’) of the positively charged imidazole are locating at 15.4 and 17.9 ppm, respectively. The downfield shifts of 4.0 ppm of HCδ2 (to 11.4 ppm, ‘H260Cδ2-II’) and 3.9 ppm of HNε2 (to 14.0 ppm, ‘H260 Nε2-II’) are observed as in the positively charged His-260 compared with its neutral form. The enhanced 1H chemical shifts (in the ω 1-dimension) also allow us to assign the doubling of the W1-signal (‘W1-I’ vs. ‘W1-II’, indicated by horizontal dashed lines A and B, respectively) with two resonances separated by ~0.6 ppm (see C1 slice in Fig. 3a and C10 in Fig. 3c). In addition, the double salt bridge between B-ring propionate (C83, Fig. 3a) and Arg-254 (two O···N distances of 2.67 and 3.33 Å, respectively) reflected as two correlations (‘R254 Nη1’ and ‘R254 Nη2’) with σ H of 8.5 and 10.8 ppm, respectively. The 2.3-ppm Δσ H for these two protons of the guanidium moiety clearly demonstrates that the Nη2 position holds a substantial portion of the positive charge in the Pr state.

2D 1H–13C interfacial correlation spectrum from u-[13C,15N]-PCB-Cph1Δ2 in the Pr state with a 2.3-ms LG-CP mixing time. Four characteristic regions are shown a–d, a carbonyl/carboxylate region (160–185 ppm), b two D-ring pyrrolic carbons (C17 and C18, as shown in the region of 130–145 ppm), c methine bridge of rings B and C (C10, as shown in the region of 103–117 ppm) and d aliphatic region (0–32 ppm). The notation for assignment uses Cph1 numbering of residues [52], followed by the Greek letter referring to the proton identity. The horizontal dashed lines mark the heteronuclear coherences originating from the same proton (with different chromophore carbons), and assist in assigning the overlapping correlation peaks
Nearly 60% of the NMR contacts involving 1H nuclei bound to the residues within ~5.5 Å of the PCB chromophore can match the intermolecular distance constraints extracted from the Cph1 2VEA Pr structure (manually counting) [52]. Part of the long-range heteronuclear correlations involving transfer of 1H polarization from methine groups (i.e., Hα, Hζ, etc.) to the carbonyls (C1 and C19, Fig. 3a as well as 10/15-methine (Fig. 3c) and the rapidly rotating methyls (C71, C171 and C182, Fig. 3d) are not resolved. This results from the truncation of weak long-range heteronuclear dipolar interactions by strong one-bond heteronuclear couplings [30, 32, 53, 54]. Despite the dominant contribution of 1H magnetization to a given 13C atom originates from its direct-bonded 1H nucleus which are completely suppressed by REDOR dipolar-filtering approach, both weak and strong heteronuclear dipolar couplings are still present in the Hamiltonian of the 1H spins, and thus the weak couplings are truncated [26, 27]. In contrast, some protons in protonated ammonium and methyl groups of nearby amino acids are found to transfer magnetization over relatively long distances, for example, Met-174 HCε···C18, 4.65 Å (Fig. 3b) and Tyr-257 HN···C81, 4.93 Å (Fig. 3d). Finally, it should be noted that we cannot identify a part of the interfacial correlations due to the inherent residual 1H resonance overlap and the 2VEA structure represents the less charge-separated ion-pair isoform only.
4 Conclusions
In this paper, we introduced a simple method for exploring long-range interfacial 1H–13C correlations between the uniformly 13C-labeled chromophore with its protein surrounding (u-[13C,15N]-PCB-Cph1Δ2), affording highly resolved 1H spectra in ω 1-dimension (e.g., the assignment of the doubling of W1-signal), which was achieved through the use of windowed CRAMPS technique (wPMLG3+) during t 1 evolution. The data are in line with the Cph1 2VEA Pr structure but permit in addition a detailed description of charge and proton dynamics within the pocket. We distinguish two co-existing isoforms of the Pr state identified by hydrogen-bonding networks and charge distributions (e.g., the proton shift of conserved His-260 in close vicinity of the chromophore). We demonstrate that this type of experiment is particularly informative for the collection of intermolecular distance restraints in selectively isotope-labeled proteins which are refractory to crystallization, such as plant phytochromes. The correlation data alone allow amino acid type assignment and, in some cases, can serve as useful fingerprints to reveal specific chromophore–protein interactions.
5 Outlook
Further efforts for improvement in terms of 1H spectral artifacts and resolution can be made by integrating a windowed 1H supercycle decoupling scheme in the form of \( wPMLG3^{ \pm } w\overline{PMLG} 3^{ \pm } \) over t 1 [18]. As the long-range 1H magnetization transfer variant of MELODI–HETCOR, selective interface detection spectroscopy (SIDY) [55] has been shown to be capable of archiving the interfacial 1H–13C contacts up to 7 Å, by means of inserting a 1H–1H spin diffusion segment at the end of the t 1 encoding period. The combination of \( wPMLG3^{ \pm } w\overline{PMLG} 3^{ \pm } \) block with the SIDY will yield interfacial HETCOR spectra, emphasizing the long-range correlations with more efficient suppression of 1H homonuclear dipolar interaction.
References
F. Castellani, B. van Rossum, A. Diehl, M. Schubert, K. Rehbein, H. Oschkinat, Nature 420, 98–102 (2002)
S.G. Zech, A.J. Wand, A.E. McDermott, J. Am. Chem. Soc. 127, 8618–8626 (2005)
A. Lange, S. Becker, K. Seidel, K. Giller, O. Pongs, M. Baldus, Angew. Chem. Int. Ed. 44, 2089–2092 (2005)
T.I. Igumenova, A.J. Wand, A.E. McDermott, J. Am. Chem. Soc. 126, 5323–5331 (2004)
W.T. Franks, D.H. Zhou, B.J. Wylie, B.G. Money, D.T. Graesser, H.L. Frericks, G. Sahota, C.M. Rienstra, J. Am. Chem. Soc. 127, 12291–12305 (2005)
T. Rohmer, C. Lang, J. Hughes, L.-O. Essen, W. Gärtner, J. Matysik, Proc. Natl. Acad. Sci. USA 105, 15229–15234 (2008)
S.J. Opella, F.M. Marassi, Chem. Rev. 104, 3587–3606 (2004)
A. Lange, K. Giller, S. Hornig, M.-F. Martin-Eauclaire, O. Pongs, S. Becker, M. Baldus, Nature 440, 959–962 (2006)
C. Wasmer, A. Lange, H. van Melckebeke, A.B. Siemer, R. Riek, B.H. Meier, Science 319, 1523–1526 (2008)
R. Siegel, J. Rocha, L. Mafra, Chem. Phys. Lett. 470, 337–341 (2009)
K. Schmidt-Rohr, H.W. Spiess, Multidimensional Solid-State NMR and Polymers (Academic Press, San Diego, 1994)
A. Bielecki, A.C. Kolbert, M.H. Levitt, Chem. Phys. Lett. 155, 341–346 (1989)
B.-J. van Rossum, H. Förster, H.J.M. de Groot, J. Magn. Reson. 124, 516–519 (1997)
E. Vinogradov, P.K. Madhu, S. Vega, Chem. Phys. Lett. 314, 443–450 (1999)
E. Vinogradov, P.K. Madhu, S. Vega, J. Chem. Phys. 115, 8983–9000 (2001)
E. Vinogradov, P.K. Madhu, S. Vega, Chem. Phys. Lett. 354, 193–202 (2002)
L. Bosman, P.K. Madhu, S. Vega, E. Vinogradov, J. Magn. Reson. 169, 39–48 (2004)
M. Leskes, P.K. Madhu, S. Vega, J. Chem. Phys. 125, 124506 (2006)
A. Lesage, D. Sakellariou, S. Hediger, B. Eléna, P. Charmont, S. Steuernagel, L. Emsley, J. Magn. Reson. 163, 105–113 (2003)
P.K. Madhu, E. Vinogradov, S. Vega, Chem. Phys. Lett. 394, 423–428 (2004)
S.P. Brown, A. Lesage, B. Elena, L. Emsley, J. Am. Chem. Soc. 126, 13230–13231 (2004)
I. Schnell, S.P. Brown, H.Y. Low, H. Ishida, H.W. Spiess, J. Am. Chem. Soc. 120, 11784–11795 (1998)
B. Reif, C.P. Jaroniec, C.M. Rienstra, M. Hohwy, R.G. Griffin, J. Magn. Reson. 151, 320–327 (2001)
I. Schnell, H.W. Spiess, J. Magn. Reson. 151, 153–227 (2001)
S.P. Brown, I. Schnell, J.D. Brand, K. Müllen, H.W. Spiess, J. Am. Chem. Soc. 121, 6712–6718 (1999)
X.L. Yao, K. Schmidt-Rohr, M. Hong, J. Magn. Reson. 149, 139–143 (2001)
X.L. Yao, M. Hong, J. Biomol. NMR 20, 263–274 (2001)
P. Caravatti, L. Braunschweiler, R.R. Ernst, Chem. Phys. Lett. 100, 305–310 (1983)
T. Gullion, J. Schaefer, J. Magn. Reson. 81, 196–200 (1989)
B.-J. van Rossum, C.P. de Groot, V. Ladizhansky, S. Vega, H.J.M. de Groot, J. Am. Chem. Soc. 122, 3465–3472 (2000)
B.J. van Rossum, E.A.M. Schulten, J. Raap, H. Oschkinat, H.J.M. de Groot, J. Magn. Reson. 155, 1–14 (2002)
J. Brus, A. Jegorov, J. Phys. Chem. A 108, 3955–3964 (2004)
C. Coelho, J. Rocha, P.K. Madhu, L. Mafra, J. Magn. Reson. 194, 264–282 (2008)
H.M. Strauss, J. Hughes, P. Schmieder, Biochemistry 44, 8244–8250 (2005)
T. Lamparter, B. Esteban, J. Hughes, Eur. J. Biochem. 268, 4720–4730 (2001)
D. Mozley, A. Remberg, W. Gärtner, Photochem. Photobiol. 66, 710–715 (1997)
L. Mafra, C. Coelho, R. Siegel, J. Rocha, J. Magn. Reson. 197, 20–27 (2009)
H. Treutlein, K. Schulten, A.T. Brünger, M. Karplus, J. Deisenhofer, H. Michel, Proc. Natl. Acad. Sci. USA 84, 75–79 (1992)
G. Metz, X. Wu, S.O. Smith, J. Magn. Reson. A 110, 219–227 (1994)
A.E. Bennett, C.M. Rienstra, M. Auger, K.V. Lakshmi, R.G. Griffin, J. Chem. Phys. 103, 6951–6958 (1995)
V.E. Zorin, M. Ernst, S.P. Brown, P. Hodgkinson, J. Magn. Reson. 192, 183–196 (2008)
J.C. Lagarias, H. Rapoport, J. Am. Chem. Soc. 102, 4821–4828 (1980)
J. Hahn, H.M. Strauss, F.T. Landgraf, H.F. Gimenèz, G. Lochnit, P. Schmieder, J. Hughes, FEBS J. 273, 1415–1429 (2006)
B. Borucki, D. von Stetten, S. Seibeck, T. Lamparter, N. Michael, M.A. Mroginski, H. Otto, D.H. Murgida, M.P. Heyn, P. Hildebrandt, J. Biol. Chem. 280, 34358–34364 (2005)
A. Remberg, I. Lindner, T. Lamparter, J. Hughes, C. Kneip, P. Hildebrandt, S.E. Braslavsky, W. Gärtner, K. Schaffner, Biochemistry 36, 13389–13395 (1997)
K. Heyne, J. Herbst, D. Stehlik, B. Esteban, T. Lamparter, J. Hughes, R. Diller, Biophys. J. 82, 1004–1016 (2002)
W. Rüdiger, F. Thümmler, E. Cmiel, S. Schneider, Proc. Natl. Acad. Sci. USA 80, 6244–6248 (1983)
S.P.A. Fodor, J.C. Lagarias, R.A. Mathies, Biochemistry 29, 11141–11146 (1990)
J. Matysik, P. Hildebrandt, W. Schlamann, S.E. Braslavsky, K. Schaffner, Biochemistry 34, 10497–10507 (1995)
H. Foerstendorf, E. Mummert, E. Schäfer, H. Scheer, F. Siebert, Biochemistry 35, 10793–10799 (1996)
A.T. Ulijasz, G. Cornilescu, C.C. Cornilescu, J. Zhang, M. Rivera, J.L. Markley, R.D. Vierstra, Nature 463, 250–254 (2010)
L.-O. Essen, J. Mailliet, J. Hughes, Proc. Natl. Acad. Sci. USA 105, 14709–14714 (2008)
M.J. Bayro, M. Huber, R. Ramachandran, T.C. Davenport, B.H. Meier, M. Ernst, R.G. Griffin, J. Chem. Phys. 130, 114506 (2009)
V. Ladizhansky, E. Vinogradov, B.-J. van Rossum, H.J.M. de Groot, S. Vega, J. Chem. Phys. 118, 5547–5557 (2003)
S.R. Kiihne, A.F.L. Creemers, W.J. de Grip, P.H.M. Bovee-Geurts, J. Lugtenburg, H.J.M. de Groot, J. Am. Chem. Soc. 127, 5734–5735 (2005)
Acknowledgments
We thank Prof. Shimon Vega (Weizmann Institute of Science, van Arkel guest Professor 2010 at Universiteit Leiden) and Dr. Thierry Rohmer for stimulating discussions. Fons Lefeber, Karthick Babu Sai Sankar Gupta and Kees Erkelens are gratefully acknowledged for their support during various stages of the experiments. Prof. Huub J. M. de Groot is acknowledged for continuous interest. This work was financially supported by the Volkswagen–Stiftung (I/79979).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Song, C., Lang, C., Mailliet, J. et al. Exploring Chromophore-Binding Pocket: High-Resolution Solid-State 1H–13C Interfacial Correlation NMR Spectra with Windowed PMLG Scheme. Appl Magn Reson 42, 79–88 (2012). https://doi.org/10.1007/s00723-011-0196-6
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-011-0196-6