
Abstract
Photosystem I (PS I) mutants from the cyanobacterium Synechocystis sp. PCC 6803 bearing point mutations to the axial ligands of A0A (M688NPsaA) and A0B (M668NPsaB) were studied by high-field W-band electron paramagnetic resonance (EPR) spectroscopy. It was found that the EPR observables of PS I from the M668NPsaB mutant were virtual identical to that of the wild type (WT), and are clearly distinct from the M688NPsaA mutant. In particular, the P ·+700 decay kinetics in the M688NPsaA mutant is significantly slower than in the WT or the M668NPsaB mutant. The analysis of the out-of-phase electron–electron dipolar electron spin echo envelope modulation shows that in the M668NPsaB mutant, the estimated distance of 26.0 ± 0.3 Å agrees well with the 25.8 Å distance for the P ·+700 A ·−1A radical pair measured in the X-ray crystal structure. In the M688NPsaA mutant, two populations are found with estimated distances of 26.0 ± 0.3 and 25.0 ± 0.3 Å in a ratio of 0.7–0.3, which agree well with the 25.8 Å distance for the P ·+700 A ·−1A radical pair and the 24.6 Å distance for the P ·+700 A ·−1B radical pair measured in the X-ray crystal structure. The data confirm that under the experimental conditions employed in this work, which involve dark-adapted samples without the pre-reduction of the iron–sulfur clusters, electron transport in cyanobacterial PS I is asymmetrical at 100 K, with the majority of electron transfer taking place through the A-branch of cofactors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Photosystem I (PS I) of cyanobacteria, algae, and higher plants is a membrane-bound chlorophyll (Chl)–protein complex that mediates light-induced electron transfer across the lipid bilayer from plastocyanin (Pc) to ferredoxin (Fd). PS I is a so-called type I photosynthetic reaction center (RC), which employs low-potential iron–sulfur clusters (FA/FB) as the terminal electron acceptors. The three-dimensional structure of PS I from the thermophilic cyanobacterium Thermosynechococcus elongatus has been solved by X-ray diffraction to a resolution of 2.5 Å [1]. The monomeric PS I complex (≈300 kDa) contains 12 different protein subunits, nine of which (PsaA, PsaB, PsaF, PsaI to PsaM and PsaX) are membrane-bound, and three of which (PsaC, PsaD and PsaE) are membrane-extrinsic. The membrane-embedded core of PS I is a heterodimer of the two largest subunits, PsaA and PsaB, which along with PsaF, PsaI to PsaM and PsaX, binds 96 molecules of Chl a, 22 molecules of β-carotene, two molecules of phylloquinone (PhQ), and an interpolypeptide [4Fe–4S] cluster FX. The terminal [4Fe–4S] clusters, FA/FB, are bound to the ≈9 kD peripheral stromal subunit, PsaC.
The arrangement of the electron transport cofactors in the PsaA, PsaB, and PsaC subunits is shown in Fig. 1. The primary electron donor, P700, is comprised of a molecule of Chl a and a molecule of Chl a′ (Chl1A/Chl1B), the porphyrin planes being parallel to one another (interplanar distance, 3.6 Å) and perpendicular to the membrane plane. The X-ray diffraction analysis reveals additional domains of high electron density which are attributed to two Chl a molecules in positions corresponding to the accessory bacteriochlorophyll molecules in the RC of purple bacteria (Chl2A/Chl2B), two Chl a molecules in positions corresponding to the bacteriopheophytin molecules in the RC of purple bacteria [2–4] (Chl3A and Chl3B), and two PhQ molecules (QA and QB). Pairs of Chl and PhQ molecules are located in nearly C 2-symmetric electron transport branches A and B associated with subunits PsaA and PsaB, respectively. Branch A incorporates Chl molecules Chl1A, Chl2A, Chl3A, and PhQ A1A, whereas branch B incorporates Chl molecules Chl1B, Chl2B, Chl3B, and PhQ A1B. The spectroscopically detected PS I cofactors include P700 (the special pair of molecules Chl1A and Chl1B), A0 (one or two pairs of Chl molecules denoted as Chl2A/Chl3A and Chl2B/Chl3B), A1 (one of the two molecules of phylloquinone (A1A/A1B), and FX, FA, and FB (the three [4Fe–4S] clusters) (see Fig. 1).

Placement of electron transfer cofactors in PS I from Thermosynechococcus elongatus [1] derived from the crystallographic coordinates in the Protein Data Bank (PDB) entry 1JBO. The electron transfer chain is indicated by the arrows. Detailed description is given in the text
According to the currently accepted paradigm, the primary electron donor P700 is promoted to the excited singlet state \( {\text{P}}_{700}^{*} \) after absorbing a light quantum. This event is followed by the primary act of charge separation between \( {\text{P}}_{700}^{*} \) and the primary acceptor A0, forming the radical pair P ·+700 A ·−0 . An alternative scheme has been proposed in which the initial charge-separated state occurs between Chl2A and Chl3A and between Chl2B and Chl3B followed by migration of the hole from Chl2A and Chl2B to P700 [5]. It should be noted, however, that this scheme has been recently challenged on the basis of electrostatic calculations of the midpoint potentials of the relevant cofactors [6]. At room temperature, the electron is transferred to the PhQ(s) A1 within 50 ps and then to the iron–sulfur clusters FX and FA/FB in ≤200 ns. In the absence of an exogenous electron acceptor, the electrons on F ·−X and [FA/FB]·− recombine with P ·+700 with lifetimes of 0.5–5 and 30–100 ms, respectively (for review, see Ref. [7]). The symmetric arrangement of the cofactors, as well as the convergence of the A- and B-branches at the FX cluster, suggest that both branches may be active in electron transfer. Interestingly, in the other main two types of photosynthetic RCs, namely bacterial RCs and photosystem II (PS II), electron transfer proceeds unidirectionally along the analogous A-branch. In these two RCs, the unidirectionality of electron transfer is functionally justified, because in contrast to PS I, the primary quinone QA serves as a single electron acceptor, while the secondary quinone QB serves as a gate to accept sequentially two electrons (and two protons).
The issue of whether electron transfer in PS I utilizes the A-branch of cofactors, the B-branch of cofactors, or both, has been a topic of lively debate for the last decade (reviewed in Ref. [8]). Two different approaches have been taken to investigate this issue. One involves altering amino acids, pigments, and cofactors in the vicinity of the A1A and A1B PhQs, and the other involves altering the H-bond and the axial ligand to the A0A and A0B Chls. The mutant PS I complexes generated near the A1 and A0 sites have been studied primarily using time-resolved optical spectroscopy and electron paramagnetic resonance (EPR) spectroscopy. The advantage of EPR is its ability to reveal spin density distributions of the transient states of the electron transfer cofactors, which reflect the electronic structure and the distances between the donor–acceptor radical pairs. The disadvantage is that to obtain information from dipolar EPR experiments [9], frozen solutions must be used at temperatures well below physiological conditions.
In Synechocystis sp. PCC 6803, site-symmetric mutations (Met → Leu) of the Chl3A/Chl3B (A0) axial ligands affect almost exclusively the A-branch electron transfer when analyzed by femtosecond to picosecond time-resolved optical spectroscopy [10], by microsecond to millisecond optical spectroscopy and by X-band (9.5 GHz) time-resolved EPR (TREPR) spectroscopy at room and low temperatures [11], and by Q-band (35 GHz) TREPR spectroscopy at low temperatures [12]. The results showed that the M688LPsaA mutant exhibits: (i) slower growth rates, higher light sensitivity, and reduced amounts of PS I; (ii) a reduced yield of electron transfer from P700 to FA/FB at room temperature; (iii) an increased formation of the 3P700 excited triplet state due to P ·+700 A ·−0 recombination; (iv) a change in the intensity and shape of the electron polarization patterns of the consecutive radical pair states the consecutive radical pair states P ·+700 A ·−1A ; and P ·+700 F ·−X (v) a slowing of the A ·−0A to A1A electron transfer step from 30 to 200 ps at room temperature. Except for an accelerated rate of charge recombination between P ·+700 and [FA/FB]·−, PS I from the M688LPsaB mutant was similar to PS I from the wild-type (WT). Site-symmetric mutations (Met → Asn) of the Chl3A/Chl3B (A0) axial ligands were also prepared and studied by femtosecond to picosecond time-resolved optical spectroscopy [10]. The lifetime of P ·+700 in the M688NPsaA and M668NPsaB mutants was found to be similar to that in the M688LPsaA and M668LPsaB mutants.
To further characterize the M688NPsaA and M668NPsaB mutants, high-field W-band EPR experiments were carried out. By extending standard X-band (9.5 GHz) EPR to high-field/high-frequency techniques, significant advantages in terms of resolution and sensitivity are realized [9]. In particular, W-band EPR spectroscopy provides an excellent opportunity to distinguish and follow separately EPR signals from the ion-radicals P ·+700 and A ·−1 (see Fig. 2). Moreover, electron–electron dipolar experiments, for instance, out-of-phase electron spin echo envelope modulation (ESEEM) on spin-correlated radical pairs [13] performed at high magnetic fields, allow one to significantly simplify the data analysis and to improve the interpretation of the results. Data analysis of standard X-band ESEEM spectroscopy must include a multiparameter fit of detected Fourier-transformed Pake-type spectra [14]. Such an analysis includes possible contributions to the electron-dipolar ESEEM spectra originating from nuclear modulations, for instance, from the 14N nuclei (nuclear Larmor frequency ν n of 1.0 MHz at 9.5 GHz EPR) of P ·+700 , as well as artifacts due to incomplete excitation of the EPR spectra of both radicals in the pair. High-field EPR is virtually free of contributions from nuclear modulations (ν n(14N) = 10 MHz at 95 GHz). Furthermore, the spectral resolution in combination with the structure of the radical pair allows one to selectively detect the perpendicular dipolar frequency, which can be analyzed by including two different dipolar contributions stemming from different radical pair geometries.

The individual absorption EPR spectra of P ·+700 and A ·−1 radicals as calculated for W-band (95 GHz). The corresponding canonical g-values of both radicals [34] as well as the placement of the cofactor radicals within the PS I structure are indicated. The individual EPR spectra are normalized to the maximum amplitude
2 Materials and Methods
2.1 Cyanobacteria and Mutants
The M688NPsaA and M668NPsaB mutants were constructed in a Synechocystis sp. PCC 6803 preparation similar to that described for the M688LPsaA and M668LPsaB mutants [11]. The mutant cells were grown photomixotrophically with 5 mM glucose in liquid culture under conditions of light-activated heterotrophic growth [15]. Growth was monitored turbidometrically at 730 nm using a Cary 14 spectrophotometer modified for computerized data acquisition. The relevant sections of the psaA and psaB genes were resequenced after every large-scale growth to confirm that the Met residue had been replaced by an Asn residue. PS I trimers were isolated from thylakoid membranes using n-dodecyl-β-d-maltopyranoside (DM) and purified on two successive sucrose density gradients. The trimeric PS I complexes were present in the bottom green band and were resuspended in 50 mM Tris, pH 8.0, containing 0.03% DM and 15% glycerol at a Chl concentration of 3 mg/ml, and stored at −80°C until required.
2.2 W-band EPR Measurements
High-field EPR measurements were performed on a homebuilt W-band (95 GHz/3.4 T) multipurpose EPR spectrometer described previously [9, 16]. The spectrometer was equipped with a TE011 optical transmission cavity with unloaded quality factor Q U = 5000 (empty). The sample solutions were placed in a quartz capillary (inner diameter, 0.6 mm) transferred to the EPR probe head and after dark adaptation at room temperature for 10 min cooled down to 100 K. Light-induced electron transfer was initiated by light pulses at 532 nm (Nd:YAG laser, 5 ns pulse width, 0.5 mJ on the sample surface) or by continuous illumination at 690 nm using continuous-wave (cw) diode laser (25 mW output, ca. 10 mW on the sample surface) guided to the center of the EPR cavity through a quartz fiber with diameter of 0.8 mm.
The heterodyne microwave bridge allowed us to perform cw EPR with field modulation and lock-in detection, TREPR without field modulation but direct detection, as well as pulsed EPR experiments. The recombination kinetics was obtained by recording the short-lived EPR absorption after laser flash (532 nm) via lock-in transient detection with magnetic field modulation (30 kHz, 0.1 mT modulation amplitude). The time resolution of this detection technique was thus set to about 1 ms. The pulsed EPR measurements were performed using the Hahn-echo sequence (tp)-τ-(2tp)-τ-echo under cw light illumination or allowing for a time after laser flash, in the sequence T DAF, hν-T DAF-(t p) x,−x -τ-(2tp)-τ-echo under pulsed light illumination. The t p pulse length of the π/2 microwave (mw) pulses was generally set to 30 ns. The phase of the first mw pulse was cycled between +x, −x. The quadrature-detected echo traces, (s −y , s x ), were digitized with 500 megasamples/s (2 ns per point) by a digital oscilloscope and transferred to the computer for further evaluation. To obtain the in-phase field-swept EPR spectra, the s −y echo response traces at τ = 150 ns, corresponding to the first pulse phase settings +x and −x, were subtracted from each other, and the pure echo response, i.e., free of free-induction decay (FID) and cavity ringing signals, was integrated over the whole echo duration. The out-of-phase EPR decay profiles were evaluated from s x echo traces by integrating the echo responses over the time window, centered at the s −y echo maximum that covers 60% of the echo intensity, to optimize resolution and signal-to-noise ratio. The echo-decay traces were acquired stepping the pulse separation time, τ, with 10 ns starting from 60 ns. The delay-after-flash time T DAF was generally set to 400 ns.
2.3 Low-Temperature Optical Measurements
Transient absorbance changes in the near-infrared were measured in PS I complexes using a laboratory-built double-beam spectrophotometer and a low-temperature cryostat (Janis Research, Wilmington, MA). The excitation beam was provided by a frequency-doubled (λ = 532 nm), Q-switched Nd:YAG laser (Spectra Physics). The pulse width at half-maximum was 8 ns. The cw measuring beam was provided by a semiconductor diode laser operating at 820 nm (Melles Griot). PS I trimers isolated from Synechocystis sp. PCC 6803 were suspended at 50 μg/ml Chl in 25 mM Tris–HCl buffer, pH 8.3, 10 mM Na ascorbate, 4 μM 2,6-dichlorophenolindophenol (DPIP), and 0.04% n-dodecyl-β-d-maltoside. The sample was frozen in the dark before measurement.
3 Results
3.1 Decay Kinetics of Flash-Induced EPR Signals from P ·+700 and \( {\text{A}}_{1}^{ \cdot - } \)
Figure 3 compares the decay kinetics of the field-modulated EPR signal from P ·+700 generated by repetitive laser flashes (1 Hz repetition rate) in PS I complexes from the WT and the M688NPsaA and M668NPsaB mutants. After a fast flash-induced generation of P ·+700 a fraction of the light-induced signal decays reversibly at 100 K (the irreversibly formed P ·+700 is subtracted and hence not represented here). The kinetics of the post-illumination reduction of P ·+700 (charge recombination) could be deconvoluted into at least three components: a fast kinetic phase (≈2 ms), a slow kinetic phase (≈200 ms), and a very slow kinetic phase (≈3 s). The half-times and statistical weights of these components, which were detected in PS I from the WT and the M688NPsaA and M668NPsaB mutants, were determined from the multiexponential fitting of the P ·+700 decay curve, and are listed in Table 1. PS I complexes from the B-branch mutant (M668NPsaB) show practically the same kinetics of P ·+700 decay as that observed in PS I complexes from the WT. PS I complexes from the M688NPsaA mutant are, however, characterized by a markedly slower reduction of P ·+700 . In contrast to the WT and M668NPsaB mutant, in which the fast component of P ·+700 decay dominates (≥50–60%), the fast component comprises only 25% of the total amplitude in the M688NPsaA mutant.

Light-induced changes in the amplitude of the EPR signals from P ·+700 in PS I complexes isolated from WT cyanobacterial cells and carrying mutations M688NPsaA and M668NPsaB. Dark-adapted samples (10 min at room temperature) were cooled down to 100 K in the dark and then illuminated with a train of laser pulses (532 nm, repetition rate 1 Hz). The inset shows the decay traces in logarithmic time scale
It should be added that the fast component of the post-illumination reduction of P ·+700 (<1 ms), which can also be observed by optical and electrometrical methods [17, 18], is likely to be underestimated in the EPR experiment due to the intrinsic time limitation of the technique [9]. We point out, however, that the measured EPR kinetics of P ·+700 decay is in very good agreement with the optical measurements. To illustrate this point, let us consider Fig. 4, in which we compare the semi-logarithmic plots of the post-illumination P ·+700 decay kinetics measured at 100 K in WT PS I complexes by both optical and EPR methods. The optically measured kinetic trace reveals a variety of exponential components. Specifically, after fast initial steps of P ·+700 decay (≈500 μs, statistical weight about 60%), slower components of P ·+700 decay are present, which are practically identical with the kinetics measured by EPR. Note that the EPR-detectable portion of the post-illumination kinetics of reversible P ·+700 reduction comprises about 40% of the maximal concentration of P ·+700 determined from optical measurements (Fig. 4). The coincidence of the EPR-detectable kinetics of P ·+700 re-reduction with the relevant portion of optically measured kinetics shows that both methods provide consistent results.

Comparison of the decay kinetics of post-illumination reduction of P ·+700 in WT PS I complexes as measured by optical (dashed line) and W-band EPR (solid line) methods at 100 K
The fast component of P ·+700 decay measured by EPR (\( \tau_{1/2}^{(1)} = 2.0 \pm 0.4\,{\text{ms}} \) in the WT and M668NPsaB mutant, and \( \tau_{1/2}^{(1)} = 12 \pm 2\,{\text{ms}} \) in the M688NPsaA mutant) can be attributed without ambiguity to charge recombination of the radical pair P ·+700 A ·−1 . In Fig. 5, we compare the EPR-measured decay kinetics for the cation-radical P ·+700 and the anion-radical A ·−1 in PS I from the M668NPsaB mutant. One can see that the dominant fast component of the A ·−1 decay (≥90%) has a half-time 1.7 ± 0.3 ms, which overlaps with the characteristic time of the fast component of P ·+700 decay. In PS I from the M688NPsaA mutant, the first component of P ·+700 decay, attributed to recombination of the radical pair P ·+700 A ·−1 , is markedly slower (\( {\tau_{1/2}^{(1)} = 12 \pm 2\,{\text{ms}}} \)) than that in PS I from the WT and the M668NPsaB mutant. In contrast to the WT and M668NPsaB samples, in which the fast EPR-detectable component dominates (≥50–60%), in the M688NPsaA sample the fast component comprises only 25% of the total amplitude (note that due to the insufficient time response of the lock-in detection EPR technique, the amplitude and lifetime of the fast component may be underestimated in the EPR measurement of the WT and M668NPsaB mutant, see Fig. 4).

EPR decay kinetics acquired at the maximum of W-band EPR absorption for P ·+700 and A ·−1 radicals in the PS I complex isolated from M668NPsaB mutant cells
The slow EPR-detectable component of P ·+700 decay (\( \tau_{1/2}^{(2)} \approx 140\,{\text{ms}} \), statistical weight ≈30%) can be attributed tentatively to recombination of the radical pair P ·+700 F ·−X . This component was absent in the kinetics of the A ·−1 decay (Fig. 5), although a similar component of the P ·+700 decay (\( {\tau_{1/2}^{(2)} \approx 200\,{\text{ms}},\; \approx 34\% } \)) was observed in the P700–FX core preparation isolated from WT cells (data not shown, see Table 1 for kinetic parameters). The very slow component (\( \tau_{1/2}^{(3)} \ge 3\,{\text{s}};\; \approx 20\% \) in the WT and ≈15–17% in the M688NPsaA and M668NPsaB mutants) remains unassigned. Because the very slow component is absent in the P700–FX core preparation, it is tempting to assign it to a previously unrecognized albeit small amount of recombination from \( {\text{P}}_{700}^{ \cdot + } [{\text{F}}_{\text{A}} /{\text{F}}_{\text{B}} ]^{ \cdot - } \). However, this assignment requires confirmation by additional spectroscopic methods.
3.2 Comparison of EPR Spectra of PS I from WT, M688NPsaA and M668NPsaB Mutants
In this section, we compare field-swept pulse EPR spectra of dark-adapted PS I complexes illuminated with continuous laser light (690 nm). As shown in Fig. 6, there are no visible EPR signals attributed to P ·+700 in dark-adapted preparations of PS I from the WT and the M668NPsaB mutant. In similar PS I preparations from the M688NPsaA mutant, however, a small signal from P ·+700 is present. Illumination of dark-frozen samples with saturating, continuous light induced a partly reversible oxidation of P700. After switching off the light, the intensities of the EPR signals from P ·+700 decreased, but did not reach the initial levels that were present prior to illumination. Thus, in each case, a significant portion of the electrons transferred at 100 K cannot recombine with P ·+700 , probably due to trapping by the terminal electron acceptors, e.g., FA and FB [18]. Note that PS I from WT cells shows a higher reversibility of the EPR signal from P ·+700 characterized by the ratio ΔP/P 0 = 0.70 (see Fig. 6 for definitions) than the two mutants. For PS I complexes from the M688NPsaA and M668NPsaB mutants, reversible changes in the amplitude of the P ·+700 EPR signal were lower, ΔP/P 0 = 0.44 and 0.57, respectively.

Field-swept echo-detected W-band EPR spectra of PS I complexes isolated from WT, and M688NPsaA and M668NPsaB mutant cells. Experimental conditions: dark-adapted samples (thin line), during the illumination with continuous 690 nm light (solid line), after switching off the light (dashed line). All spectra were acquired at 100 K. The asterisk shows a reference line from the Mn2+ standard sample
In addition to the signal from P ·+700 there are changes in the EPR spectrum due to the reduction of at least one other paramagnetic species. Figure 6 shows that the illumination of PS I with continuous red light at 100 K gives rise to signals in the low-field region that could belong to a semiquinone species. Analysis of the difference spectra (‘light on’ minus ‘light off’) allows one to extract information on possible sources of the reversible EPR signal in the low-field region. Figure 7 shows difference spectra normalized either to the amplitude of the signal from P ·+700 (a) or the low-field difference signal (b). As can be seen in Fig. 7b, the EPR spectra of the light-induced semiquinone-like radicals have the same shape, which are shape typical of the anion-radical A ·−1 . As we will see below, the small amount of the A ·−1 signal in the M688NPsaA mutant correlates with the results of the measurement of the transient radical pair P ·+700 A ·−1 generated by repeated laser flashes.

Normalized difference EPR spectra (‘light on’ minus ‘light off’ spectra shown in Fig. 6). a Difference EPR spectra normalized to the amplitude of the P ·+700 EPR signal. b Difference EPR spectra normalized to the amplitude of the A ·−1 EPR signal. The thick gray line shows the calculated A ·−1 spectrum. Samples: (1) FX-core PS I complex from the WT cells; (2) and (3) PS I complexes from the M688NPsaB and M688NPsaA mutants, respectively
3.3 Out-of-phase ESEEM Study of PS I from the M688NPsaA and M668NPsaB Mutants
The out-of-phase ESEEM signal of the P ·+700 A ·−1 radical pair provides information on the magnitude of the electron–electron dipolar coupling between the radicals, thus allowing the distance between P ·+700 and A ·−1 , r PA, to be determined (for recent reviews, see Refs. [9, 19, 20]). Figure 8a compares the out-of-phase ESEEM signals of the detected 2-pulse echo-decay in PS I from the M688NPsaA mutant (solid line) and M668NPsaB (dashed line) mutant recorded at the maximum of the EPR spectrum of P ·+700 . It can be seen that in the M668NPsaB sample, the oscillations of the signal occur with a lower frequency and are less damped as compared to the M688NPsaA sample. This difference reflects structural differences between the radical pairs generated in the PS I complexes from the two mutants.

a Out-of-phase detected ESEEM signals measured at the maximum of the P ·+700 spectrum of PS I complexes from M688NPsaA (solid line) and M668NPsaB (dashed line) mutants at 100 K. The delay-after-flash time T DAF, was set to 400 ns and the laser repetition rate to 2.5 Hz. The signal from the M668NPsaB sample is scaled to the signal from the M688NPsaA sample. b Sin-FFT amplitudes of ESEEM decays for M688NPsaA (solid line) and M668NPsaB (dashed line) samples
At W-band the modulation frequency observed in the out-of-phase detected echo-decay corresponds to the perpendicular dipolar coupling frequency, ν⊥, which value depends on the distance r PA as
where ν ⊥ is measured in s−1 and r PA in units of Å. Here, we consider only the perpendicular dipolar coupling frequency ν ⊥ and not the parallel component \( \nu_{||} \). This is because the dipolar vectors directed from P ·+700 to the semiquinones \( {\text{A}}_{{1{\text{A}}}}^{ \cdot - } \) and \( {\text{A}}_{{1{\text{B}}}}^{ \cdot - } \) are oriented almost perpendicularly to the z-axes of both quinones [21].
For a detailed analysis of the dipolar coupling, we turn to the frequency domain by performing a Fourier transformation (FT) of the zero-time reconstructed sine decay traces shown in Fig. 8a. Figure 8b compares the sine fast Fourier-transform (sin-FFT) amplitudes for PS I from the M688NPsaA and M668NPsaB mutants. There is a notable difference between samples; in particular, in the M688NPsaA mutant, the line at 3 MHz reveals a shoulder shifted towards higher frequencies. This could indicate that in the M688NPsaA mutant the radical pair P ·+700 A ·−1B makes a contribution to the overall signal. Further analysis of these data allowed us to evaluate the extent of this contribution.
Figure 9 shows the high-frequency regions of the sin-FFT amplitude spectra for the M668NPsaB mutant and the M688NPsaA mutant in the presence of dithionite, which was added to the sample to increase the signal amplitude (see Sect. 3.4). For the M668NPsaB sample (Fig. 9a), the line was fitted by a Lorentzian with a peak frequency ν 0 = 2.95 MHz and a line width Δν = 0.4 MHz estimated as the half-width at half-maximum (HWHM). Figure 9b shows the deconvolution of the sin-FFT amplitude spectrum of the M688NPsaA sample into the sum of two Lorentzian curves: one with spectral parameters of the M668NPsaB sample (ν 0 = 2.95 MHz and Δν = 0.4 MHz), and the other shifted to a higher frequency by 0.4 MHz (ν 0 = 3.35 MHz, Δν = 0.4 MHz). The statistical weights of these components are 0.7 and 0.3, respectively. According to Eq. (1), spectral lines with frequencies ν 0 = 2.95 MHz and ν 0 = 3.35 MHz correspond to distances r PA = 26.0 ± 0.3 and 25.0 ± 0.3 Å, respectively, which are in a good agreement with the X-ray structural data (Fig. 10). Taking into account that the spin density distribution in the cation-radical P ·+700 is shifted towards the B-side Chl of the P700 dimer [22, 23], and assuming that spin density in the anion-radicals A ·−1A and A ·−1B is located in the center of their quinone rings, the X-ray data suggest the following center-to-center distances: \( r_{{{\text{P}}_{700} {\text{A}}_{{1{\text{A}}}} }} = 25.8\;{\text{\AA}} \) and \( r_{{{\text{P}}_{700} {\text{A}}_{{1{\text{B}}}} }} = 24.6\;{\text{\AA}} \). These values are in good agreement with our estimates of the distances, 26.0 ± 0.3 and 25.0 ± 0.3 Å, by Eq. (1).

Spectral analysis of ESEEM sin-FFT amplitudes. a The best fit of the experimental sin-FFT amplitudes (solid line) for the M668NPsaB sample to the Lorentzian line (short dashed line), positioned at 2.95 MHz and having HWHM line width of 0.4 MHz. b The experimental sin-FFT amplitudes (solid line) for the M688NPsaA sample. The long dashed line (3) represents the calculated FFT amplitudes using the sum of Lorentzian lines with the same HWHM line width of 0.4 MHz centered at 2.95 MHz, short line (1), and 3.35 MHz, dotted line (2), with the relative weights of 0.7 and 0.3, respectively

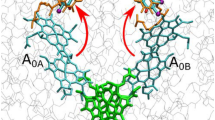
Mutual arrangement of the primary electron donor P700 (the Chl1A and Chl1B special pair) and the phylloquinones A1A and A1B based on the X-ray crystal structure data [1] (PDB entry 1JBO). The distances between P700 and A1A (25.8 Å) in the A-branch, and between P700 and A1B (24.6 Å) in the B-branch, correspond to distances between the spin density center in P700 (see Refs. [22] and [23]) and the geometrical centers of the benzoquinone fragments of A1A and A1B
3.4 EPR Spectra of the Radical Pair P ·+700 A ·−1
Next we compare the transient spin-correlated radical pair (SCRP) signals of P ·+700 A ·−1 generated by laser pulses (532 nm, 2.5 Hz repetition rate) in PS I from the WT and the M688NPsaA and M668NPsaB mutants. Figure 11 presents the SCRP signals detected 400 ns after the laser flash at 100 K. For the sake of comparison, all the spectra shown in Fig. 11 are normalized to the amplitude of the low-field component of each spectrum, which belongs to semiquinone anion radicals. As one can see in Fig. 11, the PS I complexes from the WT and the M668NPsaB mutant give rather intense signals, for which the line shapes are typical of the radical pair P ·+700 A ·−1 [9]. In the case of PS I from the M688NPsaA mutant, a much smaller SCRP signal was observed. In the presence of dithionite, which reduces FA and FB, the intensity of the signal increases by a factor of about three (not shown). This enhancement effect is probably due to an increased contribution of a B-side electron cycle as a result of the reduction of FA/FB.

Field-swept 2-pulse W-band EPR spectra of the light-induced spin-correlated radical pairs P ·+700 A ·−1 recorded 400 ns after the laser flash at 532 nm in the PS I complexes from the WT cells, and the M688NPsaA and M668NPsaB mutants at 100 K. The spectra are normalized to the A ·−1 EPR signal
It should be noted that due to the large number of factors involved, the intensity of the SCRP signal from P ·+700 A ·−1 cannot serve as a simple and unambiguous quantitative measure of the electron flux through any given branch. Nevertheless, the relatively low signal intensity in the M688NPsaA mutant, even in the presence of dithionite, is best explained either by a block in electron transfer beyond A0 in the reversible (at 100 K) A-branch of electron transfer cofactors, or by a slowing down of the recombination of the radical pairs P ·+700 A ·−1 and P ·+700 F ·−X . In the latter case, a number of active reaction centers in the state P700A1, i.e., those PS I complexes that would be in the ground state and, hence, ready for repetitive flash-induced generation of P ·+700 A ·−1 , would diminish.
The line shape of the low-field signal from A ·−1 is the same in PS I from the WT and the M668NPsaB mutant, whereas the signal in the high-field region, which is dominated by P ·+700 , differs markedly. This situation is somewhat similar to that described by Poluektov and collaborators [21], who observed at 100 K two kinds of high-frequency/high-field EPR spectra of the radical pairs P ·+700 A ·−1 in PS I complexes from predeuterated cells of the cyanobacterium Synechococcus lividus. One spectrum can be ascribed to the SCRP signal P ·+700 A ·−1A from the A-branch, while the other, observed under strongly reducing conditions, can be ascribed to the SCRP signal P ·+700 A ·−1 from the B-branch. We note, however, that the difference between the SCRP spectra, which we observed for PS I complexes from the WT and the M668NPsaB mutant and from PS I complexes from the M688NPsaA mutant, is not as dramatic as that reported in Ref. [21].
4 Discussion
The aim of the present study is to characterize PS I mutants, bearing point mutations to the axial ligands of A0A and A0B, with the use of a variety of the W-band EPR techniques. They include cw EPR, field-swept ESE, TREPR, and out-of-phase ESEEM of the spin-correlated radical pair P ·+700 A ·−1 . The use of W-band EPR afforded an opportunity to measure the kinetics of the A ·−1 radical separately from the P ·+700 radical. The results are summarized below:
-
1.
The flash-induced kinetics of P ·+700 decay coincides in PS I from the WT and M668NPsaB mutant, and differs significantly from the M688NPsaA mutant as do the relative amounts of the fast, slow and very slow components. The \( {\text{P}}_{700}^{ \cdot + } \) decay kinetics in the case of the M688NPsaA mutant is significantly slower than in the WT or the M668NPsaB mutant.
-
2.
Under continuous illumination, there is a significant difference in the amount of reversibly formed semiquinone A ·−1 between the WT, M688NPsaA and M668NPsaB samples. The amount of A ·−1 was the largest in the WT, lower in the M668NPsaB mutant, and negligible in the M688NPsaA mutant.
-
3.
The out-of-phase ESEEM study reveals a lower frequency of the spin-echo envelope modulations in the M668NPsaB mutant as compared to the M688NPsaA mutant. The analysis of these data allowed the distances between the P ·+700 A ·−1 radical pairs in the M688NPsaA and M668NPsaB mutants to be determined as 25.0 ± 0.3 and 26.0 ± 0.3 Ǻ, respectively. These distances are close to the distances estimated on the basis of the X-ray structure and spin density distribution in the P700 dimer (25.8 Ǻ for the P ·+700 A ·−1A radical pair and 24.6 Ǻ for the P ·+700 A ·−1B radical pair).
-
4.
The spin-correlated \( {\text{P}}_{700}^{ \cdot + } {\text{A}}_{1}^{ \cdot - } \) radical pair spectra were nearly identical for the WT and M668NPsaB samples, whereas for the M688NPsaA sample in the high-field region the shape of the spectrum was clearly different. In addition, the intensity of the signal in case of PS I from the M688NPsaA mutant was significantly lower than that of PS I from the WT and the M688NPsaB mutant.
The question is why the A- and B-side behaviors are so different in the two otherwise symmetrical mutants. An attempt of modeling the electron transfer rates suggests nearly equivalent midpoint potentials for A1A (−671 mV), A1B (−696 mV), and FX (−680 mV) [24]. In this model, electron transfer from A1A to FX would be endothermic by 9 mV and electron transfer from A1B to FX would be exothermic by 16 mV. The thermodynamically unfavorable electron transfer from A1A to FX at low temperature could explain the observation of the TREPR-detected P ·+700 A ·−1A radical pair on repeated laser flashes. It should be noted that the absolute values of the midpoint potentials in this study are not supported by electrostatic calculations, in which the redox potentials for A1A and A1B were calculated to be −531 and −686 mV, respectively [25]. However, Ishikita and Knapp [25] did not take into account the difference between the reference redox potential of menaquinone in water and dimethylformamide, and therefore have overestimated the absolute values of midpoint potentials for A1A and for A1B. This error does not affect the estimation of a 155 mV energy difference between A1A and A1B because the error in the liquid junction potential is the same for A1A and A1B. More recent electrostatic calculations of redox potentials, carried out within the framework of a semi-continuum dielectric approach, show that the midpoint potential of A1A is 170 mV more positive than that of A1B, and an energy gap between A1A and FX of ~80 mV [6]. The decrease in the distance typical for the P ·+700 A ·−1A pair to the distance typical for the P ·+700 A ·−1B pair, as observed by Poluektov and coworkers [21] and by Santabarbara and coworkers [26, 27], may be explained by a block in electron transfer through the A-branch. This is probably a result of reduction of A1A with dithionite and illumination (note that the more reducing A1B cofactor remains oxidized). Similarly, the observation of a change in the high-field region along with the appearance of a higher frequency shoulder in the dipolar spectrum (this work) may be explained by a partial block in electron transfer through the A-branch due to the change in the ligand to A0A and the uncovering of the relatively small amount of electron transfer through the B-branch. It remains to be explained how the otherwise fast electron transfer through the B-side cofactors can be observed within the limited EPR time window [20] even at 100 K, given that there is little or no activation energy associated with this electron transfer step [28]. Perhaps, because Asn can potentially form an H-bond to the second C=O of the quinone, the position of the A-branch quinone could have moved in some of the PS I complexes to a position closer to P ·+700 . The spectra, however, argue against this possibility because the more equal H-bonding scheme would be expected to reduce the hyperfine coupling from the –CH3 fragment.
It is clear that in Synechocystis sp. PCC 6803, the measured parameters of PS I from the M668NPsaB sample are virtually identical to those of the WT, and are clearly distinct from those that of the M688NPsaA sample. Previously, mutant PS I complexes, in which M688 on the A-branch and M668 on the B-branch were replaced by Leu residues, were studied by time-resolved optical absorption spectroscopy at room temperature [10], by X-band TREPR at room and at low temperatures [11], and by Q-band TREPR at low temperatures [12]. The measured parameters of PS I from the M668LPsaB sample were almost identical to those that of the WT, and were clearly distinct from the M688LPsaA sample. In particular, the quantum yield of electron transfer was lower, and the rate of forward electron transfer from A ·−0A to A1A was slowed by a factor of 3–10 in the M688LPsaA mutant [10]. Thus, these earlier results are consistent with the present study.
In contrast to Synechocystis sp. PCC 6803, the site-symmetric mutants M688HPsaA and M668HPsaB in C. reinhardtii showed similar effects when the decay of the electron spin-polarized signal from the P ·+700 A ·−1 radical pair was measured by low-temperature EPR. In particular, the M688HPsaA mutant showed no evidence for electron transfer to A1A at low temperature, and the M668HPsaB mutant showed no evidence for electron transfer to A1B at low temperature [29]. In a point dipole approximation, the P ·+700 -to-\( {\text{A}}_{{1{\text{B}}}}^{ \cdot - } \) distance of the M688HPsaA mutant was estimated to be 24.3 Å, and the P ·+700 -to-\( {\text{A}}_{{1{\text{A}}}}^{ \cdot - } \) distance in the M668HPsaB mutant was estimated to be 25.4 Å. The difference in the two distances is in agreement with the difference in the P700-to-A1A and P700-to-A1B distances according to the X-ray diffraction data. An intermediate value of 25.0 Å was obtained in the WT membranes, which was offered as evidence for bidirectional electron transfer [26]. Ultrafast transient absorbance measurements showed that the A0 − minus A0 difference signal accumulates in both the M688HPsaA and M668HPsaB mutants, indicating that electron transfer from A ·−0 to A1 is blocked or slowed on the appropriate branch [30]. However, a recent spin-polarized EPR study shows a transient EPR spectrum, probably due to P ·+700 A ·−1A , in the M688HPsaA variant along with no evidence for slowing of electron transfer to FX in the M668HPsaB variant [31, 32]. Indeed, a more recent time-resolved optical study of the M688HPsaA or M668HPsaA variants showed unchanged lifetimes of the fast and slow kinetic phases but an alteration in the ratio of fast to slow kinetic phases [33]. Thus, the situation in C. reinhardtii is more complex than originally thought. Further resolution of the differences and discrepancies between and among the C. reinhardtii and Synechocystis sp. PCC 6803 mutants will require additional study, possibly comparative studies of the mutants under similar conditions using the same methodologies. A paper by van der Est and colleagues [34] in this special issue further describes the low-temperature behaviour of the M688NPsaA and M668NPsaB mutants by multifrequency time-resolved and cw EPR.
References
P. Jordan, P. Fromme, H.T. Witt, O. Klukas, W. Saenger, N. Krauß, Nature 411, 909 (2001)
J. Deisenhofer, O. Epp, I. Sinning, H. Michel, J. Mol. Biol. 246, 429 (1995)
J.P. Allen, G. Feher, T.O. Yeates, H. Komiya, D.C. Rees, Proc. Natl. Acad. Sci. USA 84, 6162 (1987)
J.P. Allen, G. Feher, T.O. Yeates, H. Komiya, D.C. Rees, Proc. Natl. Acad. Sci. USA 84, 5730 (1987)
M.G. Müller, J. Niklas, W. Lubitz, A.R. Holzwarth, Biophys. J. 85, 3899 (2003)
V.V. Ptushenko, D.A. Cherepanov, L.I. Krishtalik, A.Y. Semenov, Photosynth. Res. 97, 55 (2008)
K. Brettel, W. Leibl, Biochim. Biophys. Acta 1507, 100 (2001)
N. Srinivasan, J. Golbeck, Biochim. Biophys. Acta 1787, 1057 (2009)
K. Möbius, A. Savitsky, High-Field EPR Spectroscopy on Proteins and their Model Systems: Characterization of Transient Paramagnetic States (RSC Publishing, London, 2009)
N. Dashdorj, W. Xu, R.O. Cohen, J.H. Golbeck, S. Savikhin, Biophys. J. 88, 1238 (2005)
R.O. Cohen, G. Shen, J.H. Golbeck, W. Xu, P.R. Chitnis, A.I. Valieva, A. van der Est, Y. Pushkar, D. Stehlik, Biochemistry 43, 4741 (2004)
K.M. Salikhov, Y.N. Pushkar, J.H. Golbeck, D. Stehlik, Appl. Magn. Reson. 24, 467 (2003)
R. Bittl, S.G. Zech, Biochim. Biophys. Acta 1507, 194 (2001)
C.R. Timmel, C.E. Fursman, A.J. Hoff, P.J. Hore, Chem. Phys. 226, 271 (1998)
S.L. Anderson, L.J. McIntosh, Bacteriology 173, 2761 (1991)
K. Möbius, A. Savitsky, A. Schnegg, M. Plato, M. Fuchs, Phys. Chem. Chem. Phys. 7, 19 (2005)
I.R. Vassiliev, Y.S. Jung, M.D. Mamedov, A.Y. Semenov, J.H. Golbeck, Biophys. J. 72, 301 (1997)
E. Schlodder, K. Falkenberg, M. Gergeleit, K. Brettel, Biochemistry 37, 9466 (1998)
D. Stehlik, in Photosystem I. The Light-Driven Plastocyanin:Ferredoxin Oxidoreductase, vol. 24, ed. by J. Golbeck (Springer, Dordrecht, 2006), p. 361
A. van der Est, Photosynth. Res. (2009, in press). doi:10.1007/s11120
O.G. Poluektov, S.V. Paschenko, L.M. Utschig, K.V. Lakshmi, M.C.J. Thurnauer, Am. Chem. Soc. 127, 11910 (2005)
W. Lubitz, in Advances in Photosynthesis (Series editor Govindjee). Photosystem I. The Plastocyanin: Ferredoxin Oxidoreductase in Photosynthesis, vol. 24, ed. by J.H. Golbeck (Kluwer Academic Publishers, Dordrecht, 2006), p. 245
M. Plato, N. Krauss, P. Fromme, W. Lubitz, Chem. Phys. 294, 483 (2003)
S. Santabarbara, P. Heathcote, M.C. Evans, Biochim. Biophys. Acta 1708, 283 (2005)
H. Ishikita, E.W.J. Knapp, Biol. Chem. 278, 52002 (2003)
S. Santabarbara, I. Kuprov, W.V. Fairclough, S. Purton, P.J. Hore, P. Heathcote, M.C. Evans, Biochemistry 44, 2119 (2005)
S. Santabarbara, I. Kuprov, P.J. Hore, A. Casal, P. Heathcote, M.C. Evans, Biochemistry 45, 7389 (2006)
R. Agalarov, K. Brettel, Biochim. Biophys. Acta 1604, 7 (2003)
W.V. Fairclough, A. Forsyth, M.C. Evans, S.E. Rigby, S. Purton, P. Heathcote, Biochim. Biophys. Acta 1606, 43 (2003)
V.M. Ramesh, K. Gibasiewicz, S. Lin, S.E. Bingham, A.N. Webber, Biochemistry 43, 1369 (2004)
A. van der Est, in Phototosystem I. The Light-Induced Plastocyanin:Ferredoxin Oxidoreductase, vol. 24, ed. by J. Golbeck (Springer, Dordrecht, 2006), p. 387
M. McConnell, PhD thesis, Arizona State University, Arizona, USA, 2008
F. Rappaport, B.A. Diner, K. Redding, in Photosystem I. The Light-Induced Plastocyanin:Ferredoxin Oxidoreductase, vol. 24, ed. by J. Golbeck (Springer, Dordrecht, 2006), p. 223
A. van der Est, S. Chirico, I. Karyagina, R. Cohen, G. Shen, J.H. Golbeck, Appl. Magn. Reson. doi:10.1007/s00723-009-0043-1
Acknowledgments
We are grateful to Art van der Est for fruitful discussions and for a critical reading of the manuscript. This work was supported by the Deutschen Forschungsgemeinschaft (DFG) (MO 132/19-2), the DFG-Russian Foundation for Basic Research (Cooperation Project NNIO_a 08-04-91951), the Russian Foundation for Basic Research (Grants 09-04-01657-a, 09-04-00978-a and 07-04-01050), the Federal Agency for Science and Innovation (Grants 02.512.12.2005 and 02.512.11.2286), and the US National Science Foundation (MCB-0519743).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Savitsky, A., Gopta, O., Mamedov, M. et al. Alteration of the Axial Met Ligand to Electron Acceptor A0 in Photosystem I: Effect on the Generation of P ·+700 A ·−1 Radical Pairs as Studied by W-band Transient EPR. Appl Magn Reson 37, 85–102 (2010). https://doi.org/10.1007/s00723-009-0052-0
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-009-0052-0