
Abstract
Cellulosic fibers spun from 1,3-dialkylimidazolium ionic liquids are effectively stabilized against cellulose degradation by the addition of antioxidants, but this protective effect comes at the expense of chromophore generation from the degradation products of the stabilizers. In this study, we identified the oxidation and degradation products of four natural antioxidants, α-tocopherol, N-methyl-α-tocopheramine, propyl gallate, and hydroxytyrosol, formed upon accelerated ageing of the fibers. Ageing was performed according to standard protocols under either dry or moist conditions and the extraction was done with supercritical carbon dioxide. Chromophore formation in spinning dope, upon dry ageing and moist ageing were compared. In total, 16 different oxidation/degradation products were isolated, their structure confirmed by comprehensive analytical characterization with full NMR resonance assignment in the 1H and 13C domains as well as by comparison with authentic samples, and their formation pathways discussed. Knowledge of the chemical structures of the degradation products originating from the stabilizers now provides a good starting point for optimization of the fiber bleaching stage.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The production of man-made cellulosic fibers by spinning from cellulose solutions has not only evolved into an important facet of sustainable materials science but is above all an economically significant factor and a mainstay of the pulp-based industries. Approximately two decades ago, ionic liquids, specifically N-alkyl-N-methylimidazolium ILs, have emerged as promising solvents for cellulose and potential pulping agents. However, the initial euphoria faded with unsurmountable challenges in recycling and purification, hindering their widespread adoption. Subsequent advancements in IL technology, particularly with higher-generation ionic liquids, addressed these challenges and culminated in the development of the Ioncell® technology [1,2,3,4,5] for cellulose dissolution and fiber spinning on a larger scale. Recently, also the first-generation ILs with 3-alkyl-1-methylimidazolium structure have regained attention in the quest for eco-friendly and efficient cellulose fiber production [6, 7], applied in the production of special high-strength cellulose fibers from pure pulps which overcome the usual recycling and purification hurdles. However, the inherent reactivity of these solvents [8, 9] and the involvement of oxidative processes during dope preparation, spinning, and fiber ageing pose considerable challenges, necessitating the use of stabilizers—which is the general topic of our investigation.
Despite the successful suppression of cellulose degradation during dissolution and fiber spinning through the incorporation of antioxidants, a concomitant issue arises—the formation of chromophores from the stabilizers—leading to discoloration of the spinning dope and the resultant fibers. Previous studies have demonstrated that the degradation of cellulose upon dissolution in 1,3-dialkylimidazolium ionic liquids, formation of a spinning dope, and spinning of cellulosic fibers from this dope, is mainly due to homolytic degradation reactions, i.e., processes that involve atmospheric oxygen [10] and radical oxygen species as shown by spin trapping [11]. The addition of antioxidants, commonly 2wt% relative to the dissolved pulp, can effectively curb these cellulose degradation reactions. This stabilization, however, comes at the cost of increased discoloration of the spinning dope. The resulting decrease in brightness of the spun fibers poses challenges in terms of bleachability, chemical needs for bleaching treatments and brightness stability. To tackle this issue, a deeper understanding of the chemical structures of the generated chromophores becomes imperative. Following green chemistry principles, the focus was on stabilizers from renewable resources rather than synthetic antioxidants. Luckily, the four natural antioxidants α-tocopherol (1), N-methyl-α-tocopheramine (2), propyl gallate (3), and hydroxytyrosol (4), see Scheme 1, were among the stabilizers showing optimum overall performance in a comparison with 12 commercially available antioxidants [10].

They gave the best-balanced results with regard to minimizing side reactions in the spinning dope, minimizing cellulose degradation in the spinning dope and the spun fibers, and limiting chromophore generation in the spinning dope and resulting fibers. These four antioxidants have been studied in detail with regard to their protective effect in the spinning dope and the generation of oxidation products. The concomitant chromophore generation and discoloration can be counteracted by a fiber bleaching/washing step, an approach that is generally used in the manufacture of man-made cellulosic fibers, such as viscose (Rayon) or Lyocell fibers. In the case of the studied antioxidants, roughly one-third of the stabilizer amount is used up during dope preparation and spinning, the other two-thirds are incorporated into the fibers to prolong the shelf-life and for long-term stabilization of the cellulose.
In this study, we address the formation and identification of oxidation and degradation products of the antioxidants in the spun fibers upon long-term storage, in particular the chromophoric products. Long-term behavior is simulated by accelerated ageing under either dry or moist conditions according to standard procedures in cellulose fiber and paper science. The exact knowledge of the chemical structures of the reaction products is necessary on the one hand to exclude the formation of any unwanted harmful compounds and on the other hand to optimize the final bleaching step in the fiber treatment with regard to effectivity, mild conditions, and chemical savings.
Results and discussion
Stabilizing effect of the antioxidant additives 1–4
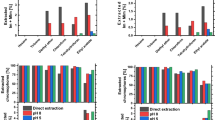
The beneficial effect of the stabilizers can be demonstrated in a particularly striking way by comparing the celluloses’ molar mass (Mw) of the stabilized fibers to a non-stabilized blank (Figs. 1 and 2). For all stabilized fibers the Mw values stayed above 85% of the initial pulp value, while there was a steep drop to about 50% in the case of the non-stabilized pulp. After 12 h of ageing, the stabilizers α-tocopherol (1), N-methyl-α-tocopheramine (2), and propyl gallate (3) had been largely consumed (see below), which is reflected by a sharper decrease of Mw in the interval between 12 and 24 h than before. In the case of hydroxytyrosol (4), there was only a minor decrease between 12 and 24 h, comparable to the ageing period before, because the stabilizer was still abundantly present after 12 h (see below) and thus still ready to act. The Mw loss of the non-stabilized fibers between 12 and 24 h of ageing was drastic, dropping to approx. 30% of the starting pulp value. There was no significant difference between dry and wet ageing in the ageing behavior and stabilizing efficiency, although some mechanisms and product distributions were well dissimilar, as discussed below.

Cellulose degradation (as molar mass loss relative to the starting pulp) upon spinning dope preparation and dry aging over different times. Toc: α-tocopherol, Toc-amine: N-methyl-α-tocopheramine, PG: propyl gallate, HyT: hydroxytyrosol, no stab.: no stabilizer used

Cellulose degradation (as molar mass loss relative to the starting pulp) upon spinning dope preparation and moist aging over different times. Toc: α-tocopherol, Toc-amine: N-methyl-α-tocopheramine, PG: propyl gallate, HyT: hydroxytyrosol, no stab.: no stabilizer used
Figures 1 and 2 show the cellulose degradation upon spinning dope preparation (5 wt% cellulose in 1-ethyl-3-methylimidazolium acetate, EMIm-OAc) and subsequent dry and moist accelerated ageing of the spun fibers, in dependence on the added stabilizers (0.1 wt% rel. to dope, 2 wt% rel. to dissolved pulp). The molar mass, determined by gel permeation chromatography according to the standard protocol [12] is expressed as molar mass loss [%] relative to the starting molar mass of the pulp: (Mw0-Mw)/Mw0 [%], with Mw being the determined molar mass and Mw0 being the starting molar mass of the pulp.
The changes in ISO brightness with ageing are currently being investigated in detail. Suffice it to say that after 12 h of ageing, a brightness loss of approx. 5–6 ISO points was observed for all stabilizers, which corresponds approximately to the value for a non-stabilized fiber. While in this non-stabilized fiber, the chromophores are formed directly as a result of the oxidation of the polysaccharide components and subsequent condensation reactions [13], the chromophores in stabilized fibers are almost exclusively degradation products of the stabilizers: after their extraction, the brightness value returned to that of the unaged fiber.
Extraction of stabilizers and their degradation/oxidation products
All stabilizers and their degradation products are strongly to moderately lipophilic compounds, which is particularly true for the tocopherol derivatives with their isoprenoid C16H33 side chains [14]. While extraction from the aqueous spinning bath was easily possible by liquid–liquid extraction into toluene (unpublished work), the degradation products in the spun and aged fibers are tightly embedded in the fiber structure and not accessible to extraction with apolar organic solvents. To increase accessibility, swelling of the fiber with a polar aprotic solvent would be required. Such swelling in N,N-dimethylacetamide is used, for instance, as pretreatment before the dissolution of cellulosic pulps for analytical purposes (gel permeation chromatography, see [12]), or swelling in DMSO in the case of particularly hard-to-dissolve cellulosic fibers [15]. However, it was questionable whether the degradation products would remain chemically unchanged in this process and whether the swelling agents would extract the apolar degradation products. The addition of an apolar solvent would not help as it could increase solubility, but at the same time would re-collapse the swollen fibers. For this reason, we resorted to extraction by supercritical carbon dioxide [16, 17], which offered the best solubility for the apolar substances and superb accessibility through the bulk of the fibers due to the extremely high diffusivity of the supercritical medium. In addition, the extract did not need any purification, drying, or evaporation treatments.
The extracts from the fibers (after 12 h of ageing) were screened by HP-TLC and GC–MS against a library of standard derivatives and oxidation products of the antioxidants used, which has resulted from previous work on antioxidants and has been expanded over the years to cover the common stabilizers used in cellulosic materials. Subsequent flash chromatography of the mixtures (toluene/n-heptane on silica gel) provided the separated major components. Extraction of the freshly spun fibers, for comparison, showed that approx. 70% of unchanged antioxidants were present. This agrees perfectly well with the values determined previously for the spinning dopes, which contained also about 70% of the antioxidants and only 30% of oxidation products. By contrast, after the fiber ageing, the antioxidants were almost completely converted into their oxidation products in the case of the tocopherol derivatives and propyl gallate, with the amount of unchanged stabilizers being below 10% and without significant differences between wet and dry ageing (Fig. 3). For hydroxytyrosol, not only the conversion rate was generally much lower, but there was also a significant gap between dry and moist ageing (74% vs. 51% consumption, respectively). As the protective effect was roughly the same for all four stabilizers (see Figs. 1 and 2), it can be concluded that hydroxytyrosol would provide a significantly longer stabilization time than the other three antioxidants when applied in the same molar amount, or it could be applied in smaller amounts to reach the same stabilization duration. In general, the continuous consumption of the stabilizers upon ageing nicely demonstrates the action of the antioxidants in the fibers and mirrors the protective effect on cellulose integrity as discussed above.

Conversion of the tested antioxidants into their oxidation products in the spinning bath and after dry or moist fiber aging
Identification of the isolated chromophores and degradation/oxidation products
All identified chromophoric reaction products were analytically characterized by their NMR spectra, MS data, and the comparison to authentic samples, which were either available from previous work or independently synthesized. The identified compounds cover more than 95 wt% of the isolated reaction products for all four antioxidants used. The residual 5 wt% of minor side products (data not given here) cannot be considered as unambiguously identified as their amount was too low for comprehensive NMR analysis, although HP-TLC and MS data provided some indication as to their identity in almost all cases.
α-Tocopherol (1) is the main component of vitamin E, a mixture of tocopherols that are distinguished by different methylation patterns at the aromatic ring, with α-tocopherol—as the permethylated derivative—being the major component. It is the most common lipophilic antioxidant in vivo and most widely used in the food, feed, and cosmetics industries as additive E 307 [18]. But also in polymer and material science, it has found many usages as a stabilizer, antioxidant, and scavenger of radicals and reactive oxygen species, both in synthetic and renewable polymers and fibers [19,20,21,22]. Upon fiber ageing, α-tocopherol was converted into three major products, para-tocopherylquinone (5) [23, 24], the spiro-dimer of α-tocopherol (6) [25, 26], and the 5,6-ortho-quinone, so-called “α-tocored” (7) [27]. The two former compounds are intensively yellow waxes in pure form, while the latter—as the name indicates—has a dark red color. It is responsible for the faint red hue of fibers stabilized with α-tocopherol. Compounds 5 and 6 are the typical reaction products of α-tocopherol, the para-quinone is the end product of oxidation in the presence of water, and the spiro-dimer is an almost ubiquitous reaction product of 1 which is formed by dimerization of the α-tocopherol-derived ortho-quinone methide (1a) [28, 29] in a hetero-Diels–Alder process with inverse electron demand. Intermediate 1a is a central compound in the reactions of vitamin E with a rich chemistry. The selective involvement of the 5a-methyl group in ortho-quinone methide formation (while the other ortho-methyl group is rather unreactive) is a peculiarity of α-tocopherol which has been early recognized and accounted for by the theory of strain-induced bond localization [29, 30].
While para-quinone and spiro-dimer were also found in the spinning dope, α-tocored (7) was not: it was only present in the aged fibers and evidently formed only later upon ageing. It can thus well be used as an indicator for long-term ageing/storage of such tocopherol-stabilized fibers. A minor antioxidant-derived component (2%) isolated from the fibers was N-(5a-α-tocopheryl)imidazole (8) which was formed by the addition of the ortho-quinone methide to traces of imidazole present. Imidazole and 1-methylimidazole are almost ubiquitous byproducts in imidazolium ionic liquids and the cellulosics processed from them [31]. The reaction of the ortho-quinone methide with nucleophiles is a common reaction path in vitamin E chemistry, although the resulting 5a-substituted tocopherols are rather synthesized by reaction of the readily accessible 5a-bromo-α-tocopherol with the respective nucleophilic co-reactants [32]. Interestingly, para-quinone 5 can form the ortho-quinone methide 1a by cyclization of an 8a-hydroxy-α-tocopherone (5a) derivative which undergoes a [1,4]-elimination of water, a reaction that has been frequently exploited in tocopherol chemistry. In the present case of fiber ageing, the reaction is responsible for the fact that upon dry ageing, spiro-dimer 6 dominated over the para-quinone 5 (72% vs. 8%) while in moist ageing the para-quinone was the dominant oxidation product (52% vs. 32%). The amount of α-tocored was 16% under both conditions and thus moisture-independent.
Scheme 2 shows the oxidation products (5–8) from the antioxidant α-tocopherol (1) in the aged fibers, spun from EMIm-OAc, and indicates their interrelation and conversions. Note that all oxidation products found are naturally occurring metabolites of α-tocopherol with well-known biocompatibility.

N-Methyl-α-tocopheramine (2) is a chemical sibling of α-tocopherol in which the phenolic OH group is replaced by an N-methyl moiety. As in the group of tocopherols, the α-homolog, i.e., the compound permethylated at the aromatic ring, is the most common representative also among the tocopheramines [33, 34]. Both tocopherols and tocopheramines are fully biocompatible and non-toxic [35] and excellent antioxidants [14] with wide applications in food and feed [36, 37], polymer technology [38, 39], and in the biomedical field [40,41,42,43,44,45]. N-Methyl-α-tocopheramine is obtained by demethylation of the N,N-dimethyl derivative [46] rather than by methylation of the parent compound α-tocopheramine, which always provides a hard-to-separate product mixture. In the cellulosic fibers stabilized with 2, three reaction products were found, with para-α-tocopheryl quinone (5) as the main component. It is formed by oxidation of 2 to the corresponding para-iminoquinone, which immediately reacts with moisture under the formation of para-α-tocopheryl quinone (5) and release of methylamine. The second main product upon ageing under dry conditions was the spiro-dimer of N-methyl-α-tocopheramine (9), accompanied by small amounts of the spiro-dimer of α-tocopherol (6), in a molar ratio (9/6) of approx. 4.5:1, determined by weight. Upon ageing under moist conditions, this situation reversed, and 6 became by far dominant over 9, with a ratio (9/6) of approx. 1:12. This demonstrates that the spiro-dimer of N-methyl-α-tocopheramine (9), once formed by dimerization of the iminoquinone methide intermediate (2a) by analogy to the α-tocopherol case, is not stable, but loses methylamine in the presence of moisture, being converted to the spiro-dimer of α-tocopherol. Note that both spiro-dimers possess a fluxational nature with the aromatic part and the spiro-keto part changing their positions, as demonstrated by Netscher [26]. This explains why both nitrogen functionalities in 9 are replaced by the equivalent oxygen functionality in the reaction with water, and not only the more accessible N-methylamino moiety.
Interestingly, while α-tocored was contained in the spinning dopes stabilized with 2, it was not found in the corresponding aged fibers, and thus was not newly formed from residual 2 upon accelerated ageing. This is a major difference to the α-tocopherol case. The absence of N-(5a-α-tocopheryl)imidazole (8) and other 5a-substituted tocopherol derivatives is in accordance with vitamin E chemistry, as the quinone methide intermediate 1a easily adds nucleophiles to give 5a-substituted α-tocopherols, while the iminoquinone counterpart 2a does not undergo such processes.
The reason for the use of the N-methyl derivative 2 as the stabilizer—instead of the non-methylated parent compound α-tocopheramine—is its well-defined oxidation chemistry, which avoids dark colored compounds and affords only innocuous products that are known as natural vitamin E metabolites. The reaction of the unmethylated tocopheramine, by contrast, leads to the intensively red-colored azo-tocopherol as the main oxidation product [47], besides minor amounts of the potentially toxic nitrophenol 5-deoxy-5-nitro-α-tocopherol [48]. The N,N-dimethyl derivative has a significantly lower stabilizing effect as an antioxidant and has therefore not been used as a fiber stabilizer. The oxidation products from the antioxidant N-methyl-α-tocopheramine (2), compounds 5, 6 (Scheme 3), are naturally occurring metabolites of α-tocopherol and are known to be non-toxic.

Propyl gallate (PG, 3) is very commonly employed as a stabilizer and antioxidant: in food and cosmetic preparations as additive E 310, but also for polymeric materials and fibers [49,50,51]. It is also used, for instance, as a stabilizer in the production of cellulose Lyocell fibers in the solvent N-methylmorpholine N-oxide [39]. The aged fibers from dopes stabilized with propyl gallate contained mainly ellagic acid (11), a yellow crystalline compound in pure form, and its bis(ortho-quinone) 12, a black amorphous solid. Both are the typical oxidation products of gallic acid and its esters. The primary phenoxyl radical intermediates formed from PG in its antioxidative action, recombine to intermediate 3,3′,4,4′,5,5′-hexahydroxydiphenic acid dipropyl ester (3a), which immediately affords ellagic acid (11) by bislactone formation. This cyclization is entropically driven and proceeds under the release of n-propanol from the ester moieties. The bis(ortho-quinone) 12, with its intensive blue-to-black color due to the highly conjugated double bond system, is chiefly responsible for the rather pronounced brightness loss of PG-stabilized fibers upon ageing.
While in the spinning dope free gallic acid was found, no gallic acid was present in the spun and aged fibers. Instead, gallic acid imidazolide (13) was found as a minor component (3%). Formation of this compound cannot involve residual gallic acid, because amide formation from free acids would not occur, but would rather proceed as aminolysis of ester 3. As mentioned above, imidazole, as the co-reacting amine component, is a common trace impurity when cellulose is processed in imidazolium ionic liquids. Scheme 4 summarizes the byproducts formed upon the ageing of PG-stabilized cellulosic fibers spun from the EMIm-OAc imidazolium ionic liquid.

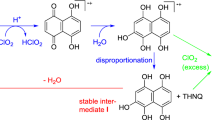
Hydroxytyrosol (4) is a main antioxidant component in olive oil and olive leaf extract and is known for its beneficial nutraceutical effects [52, 53]. Its abundant occurrence in olive mill wastewater (OMWW) has promoted also non-food applications as a stabilizer for polymers [54, 55] and fibers [56, 57]. From EMIm-OAc-spun and subsequently aged fibers, the reaction products shown in Scheme 5 have been isolated. The yellow-orange ortho-quinone (14) is the primary product of both homolytic and heterolytic oxidation. While it was the dominant reaction product in the spinning dope (short time, lower conversion), it was a minor component among the degradation products formed upon ageing (longer time, high conversion). Its intramolecular Michael addition product (14a), also found in the spinning dope, was not detected after ageing. Instead, a mixture of oxidation products with roughly equal concentrations was found (15, 16, 16a, 17, 18). Their formation pathways and interconversions will be discussed in detail in an upcoming account. Scheme 5 presents their structure and a brief formation mechanism summary.

ortho-Quinone 14 is a tautomer of para-quinone methide 14b; such ortho-quinone/para-quinone (methide) tautomerization are very well-known, for instance, in lignin [58,59,60] or vitamin E chemistry [14, 29]. The para-quinone methide immediately adds a nucleophile—most commonly water/OH−—to form a benzyl alcohol derivative under rearomatization, which is also the thermodynamic driving force for the process. The resulting 1,2-dihydroxytyrosol (15), like its parent compound hydroxytyrosol (4), is a strong antioxidant and further oxidized to the corresponding ortho-quinone 16. Similar to the ortho-quinone 14, which affords 14a, also ortho-quinone 16 undergoes intramolecular Michael addition to the bicyclic compound 16a which, in turn, undergoes elimination of water in the furan ring and rearomatization in the benzene moiety. The lack of other intermediates (compounds in which either water elimination or rearomatization had occurred, but not both at the same time) seems to indicate that both processes proceed simultaneously. The rearomatization process involves two tautomeric rearrangements, a keto-enol tautomerism (4-C = O) and a [1,5]-sigmatropic proton shift (3-C = O). The resulting dihydroxybenzofuran 17 was the major component found in the mixture of isolated oxidation products. The final product appears to be ortho-quinone 18, which is obtained by further oxidation of 17. Note that one molecule of hydroxytyrosol (4) is able to consume 6 one-electron oxidation-equivalents on the way to being eventually converted into ortho-quinone 18. This explains its high antioxidative activity and also its high effectiveness as a stabilizer in the studied fibers.
TLC screening indicated that the formation of the Michael addition products found (14a, 16a) was not an artifact of the work-up and isolation procedure, with increasing amounts being formed upon ageing. In both cases, the reaction could be reversed by dissolution in trifluoroacetic acid to generate the corresponding monocyclic ortho-quinones 14 and 16.
Comparison of the degradation product mixtures from spinning dope, dry-aged and moist-aged fibers
The composition of the mixtures of degradation products from the stabilizers shows some significant differences when comparing the chromophores in the spinning dope on one side and those in the aged fibers on the other side. By contrast, the differences between moist and dry ageing are only minor – they can be traced back to reaction steps in which water is directly involved. Table 1 summarizes the differences in the mixtures of degradation products according to a simple system that classifies the degradation products as major product (> 50%, + + +), product (+ +), and minor component (< 5%, +). In this way, also the mechanistic differences, as discussed above, are visualized.
Important differences are the occurrence of α-tocored (7) in the aged fibers stabilized with α-tocopherol, but not in the corresponding spinning dope, while for the stabilizers N-methyl-α-tocopheramine the situation is reversed: α-tocored was found only in the dope, but not in the fibers. For both tocopherol-stabilizers, moist ageing conditions favor the formation of the para-α-tocopherylquinone (5) over the spiro-dimers (6, 10) which is understandable from the formation mechanism. Oxidation to the para-quinone involves water, the formation of the spiro-dimers does not. Formation of the spiro-dimer from -tocopherol (6) by hydrolysis of the spiro-dimer from the tocopheramine (10) is a hydrolytic process that proceeds faster under moist ageing conditions than under dry ones.
The formation of 5a-(1-imidazolyl)-α-tocopherol (8) and gallic imidazolide (13) occurs only in the fibers, and not in the spinning dope. Both involve not only stabilizer-derived precursors but also imidazole as a degradation product of the ionic liquid. In the case of the stabilizer hydroxytyrosol (4), only the first oxidation product, ortho-quinone 14, is found in the spinning dope. In the fibers, both stabilizer and 14 are largely depleted by further transformation into more highly oxidized products (16–18), of which 18 seems to be the final product that is stable and not further modified.
Conclusions
In this study, we have investigated the formation of oxidation / degradation products from stabilizers in cellulosic fibers spun from the EMIM-OAc ionic liquid. All stabilizers were natural antioxidants, namely α-tocopherol, N-methyl-α-tocopheramine, propyl gallate, and hydroxytyrosol. While these antioxidants effectively protect against cellulose degradation during dope preparation, fiber spinning and fiber shelf-life, a trade-off emerges in the form of chromophore generation, leading to discoloration of the spinning dope and resulting fibers. The antioxidants’ protective effects were demonstrated by comparing the cellulose molar mass of stabilized and non-stabilized fibers, revealing a significant retention of molar mass in the stabilized fibers, highlighting the effectiveness of the chosen antioxidants. The concomitant conversion to chromophores was seen as a noticeable brightness loss upon accelerated ageing.
The study addressed the identification of oxidation and degradation products of the four antioxidants formed upon accelerated ageing of the fibers under dry and moist conditions. It employed advanced techniques, such as supercritical carbon dioxide extraction, to isolate degradation products from the aged fibers, and NMR spectroscopy with comparison to authentic samples for final unambiguous identification. A total of 16 different oxidation/degradation products were isolated, analytically characterized, structurally identified, and their formation pathways discussed. The antioxidants exhibited varying degrees of degradation, and their consumption was influenced by both the type of antioxidant and the ageing conditions. α-Tocopherol and N-methyl-α-tocopheramine were largely consumed during ageing, leading to the formation of specific chromophores such as para-tocopherylquinone, spiro-dimers, and α-tocored. Propyl gallate, also largely consumed during ageing, resulted in the generation of ellagic acid and its bis(ortho-quinone), contributing to the observed discoloration. Hydroxytyrosol, a prominent component in olives, exhibited a special effect: it formed a yellow-orange ortho-quinone as an initial oxidation product, which was further transformed into secondary products that were potential antioxidants as well. Its overall consumption rate was thus lower than in the case of the other three antioxidants. Interestingly, imidazole, as almost ubiquitous trace byproduct of the imidazolium ILs, was incorporated into the stabilizer degradation products in two cases.
The differences observed between the chromophores in the spinning dope and those in the aged fibers underscore the dynamic nature of antioxidant degradation during fiber processing and ageing. Moisture content during ageing influenced the distribution of certain chromophores, emphasizing the importance of considering environmental conditions in fiber stabilization processes (see Table 1).
The results provide valuable insights into the chemical structures of the chromophores generated during the ageing process. This knowledge serves as a crucial foundation for optimizing the fiber bleaching stage, addressing challenges related to brightness loss, chemical needs for bleaching treatments and brightness stability. Alternative antioxidants, which meet the principles of green chemistry but at the same time do not produce chromophoric reaction products upon their action, are evidently also an important direction of research. Future research will now focus on refining the stabilizer dosage, exploring alternative stabilization methods, and optimizing the bleaching process to enhance the commercial viability and sustainability of cellulosic fibers spun from 1,3-dialkylimidazolium ILs.
Experimental
All chemicals were of the highest purity available and used without further purification. Bidistilled water was used for all aqueous solutions, extractions, and washing steps. 1,4-Dioxane, n-heptane, ethyl acetate, and toluene used in chromatography were distilled before use. TLC was performed using Merck silica get 60 F254 pre-coated plates, and flash chromatography on Baker silica gel (40 µm particle size). All products were purified to homogeneity by TLC / GC analysis and satisfying elemental analysis data (± 0.2%). Elemental analyses were performed at the Microanalytical Laboratory of the University of Vienna. The melting points are corrected (benzophenone 48–49 °C, benzoic acid 122–123 °C), determined on a Kofler-type micro hot stage with Reichert-Biovar microscope.
The same cellulosic pulp, having an α-cellulose contents of 97.5% according to the manufacturer, and fibers produced thereof was used as in previous work [6, 7], Mn = 99.3 kDa, Mw = 362.7 kDa, degree of polymerization (DP) = 2237, dispersity Ɖ = Mw/Mn = 3.65, and ISO brightness = 82. The molar mass data were determined by size exclusion chromatography according to the published standard protocol [12]. The EMIm-OAc ionic liquid was obtained from Nippon Nyukazai Co., Ltd. (Tokyo, Japan).
Gel permeation chromatography (size exclusion chromatography) was used applying the standard protocol according to Potthast et al. [12], where also the SEC-MALS system is described in detail. In short, DMAc-activated samples were placed in 4 cm3 vials, and an N,N-dimethylacetamide (DMAc)/lithium chloride solution (1 cm3, 9% w/v) was added to each sample. The vials were vortexed for 20 s and placed in a rotary shaker again for over 20 h to dissolve the samples, then the samples were diluted 1:3 (v/v) with pure DMAc and filtered through PTFE syringe filters (0.45 µm) before SEC-MALS analysis. The calculations were based on a refractive index increment for cellulose of 0.136 cm3 g−1. Viscometry measurements used the solvent cupriethylene diamine (cuen) according to a previously published, improved protocol [61], which avoids systematic underestimation errors due to β-elimination reactions [62].
GC–MS/FID and UPC2-ESI-QTof-MS analysis were performed as previously described by Barbini et al. [16, 17]. UV/Vis spectra were recorded on a LAMBDA 45 UV/Vis spectrophotometer (Perkin Elmer, Waltham, MA, USA): range of 400 to 700 nm, scanning speed 480 nm min−1, quartz glass cuvettes (l = 1.0 cm).
Preparation of spinning dopes and dry-jet wet spinning
The cellulosic pulps were dried at 100 °C for 3 h. In a stainless-steel vessel, a pulp aliquot (5 wt% rel. to the IL solvent) was added to 1-ethyl-3-methylimidazolium acetate ionic liquid containing the stabilizer (2 wt% rel. to the mass of cellulosic pulp) and stirred at room temperature for 1 h. The mass of added stabilizer corresponds to 0.76 mol% for α-tocopherol (1), 0.74 mol% for N-methyl-α-tocopheramine (2), 1.56 mol% for propyl gallate (3), and 2.15 mol% for hydroxytyrosol (4), relative to cellulose (molar mass of the gluocopyranose unit: 162.135 g mol−1). The mixture was heated to 100–130 °C under stirring to affect the dissolution of the pulp. Complete dissolution, occurring after an additional 1–2 h, was confirmed using a polarized optical microscope (Olympus BHT-P, Tokyo, Japan) under crossed Nicols.
Spinning was carried out according to a dry-jet wet spinning process as reported previously [6, 7, 10]. At a temperature of 100 °C, the spinning dopes were extruded through the spinning nozzles into an air gap (15 cm) and distilled water (without any additives) as the coagulation bath, at a constant throughput speed of 0.1 cm3 min−1 and a constant winding speed of 107 m min−1. The resulting fibers were washed with hot distilled water (70 °C) to remove the major part of the solvent and byproducts, loosely coiled on bobbins, agitated in water overnight to remove residues of solvent and byproducts, and dried at room temperature.
Accelerated ageing of the fibers
Hand sheets were prepared from 2 g of fibers suspended in distilled water (500 cm3) on a Büchner funnel, followed by pressing and drying at 92 °C for 5 min. Brightness was measured before and after ageing according to ISO 2470 (2009) [63], detecting UV/Vis remission at 457 nm. Continuous ageing was performed either under dry conditions according to the TAPPI method UM 200 (105 °C, 40% rel. humidity, 4 h, 12 h, 24 h) [64] or under humid conditions according to Paptac E.4P (100 °C, 100% rel. humidity, 4 h, 12 h, 24 h) [65]. The ageing progress was monitored by UV/Vis (brightness reversion) to follow the kinetics of chromophore generation.
Extraction of stabilizer-derived degradation products and chromophores from the spinning baths by supercritical CO2
Supercritical extraction of the aged fibers was carried out with carbon dioxide (> 99.5% food grade, Biogon C, Linde AG, Austria) on a high-pressure SF-1 supercritical fluid system (Separex, Champigneulles, France) equipped with preheater, autoclave, and separator. Aliquots of the fibers (approx. 25 g) were extracted for 90 min at a flow rate of 30 g min−1, and the extracts were combined. Extraction proceeded at 30 MPa (300 bar) and 40 °C (ρ CO2: 910 kg m−3) in a cylindrical extraction vessel (diameter: 35 mm, height: 50 mm). In the case of propyl gallate-stabilized fibers, 1 wt% of ethanol 96 vol% (ρ20°C: 808.5 kg m−3, Merck) was added as a co-solvent by an HPLC pump (Lab Alliance) to enhance the solubility of ellagic acid (11) and its bis(ortho-quinone) (12). The extracts were stored under argon gas at –20 °C in brown-glass vials.
NMR analysis and identification of chromophores
All NMR spectra were recorded on a Bruker Avance II 400 (resonance frequencies 400.13 MHz for 1H and 100.63 MHz for 13C) equipped with a 5 mm N2-cooled cryoprobe head (Prodigy) with z-gradients at room temperature with standard Bruker pulse programs. The sample was dissolved in 0.6 cm3 of CDCl3 (99.9% D). Chemical shifts are given in ppm, referenced to residual solvent signals. 1H NMR data were collected with 32 k complex data points and apodized with a Gaussian window function (lb = − 0.3 Hz and gb = 0.3 Hz) prior to Fourier transformation. 13C spectrum with WALTZ16 1H decoupling was acquired using 64 k data points. Signal-to-noise enhancement was achieved by multiplication of the FID with an exponential window function (lb = 1 Hz). All two-dimensional experiments were performed with 1 k × 256 data points, while the number of transients (2–16 scans) and the sweep widths were optimized individually. HSQC experiment was acquired using adiabatic pulse for inversion of 13C and GARP-sequence for broadband 13C-decoupling, optimized for 1 J(CH) = 145 Hz. For the NOESY spectra, a mixing time of 0.8 s was used.
The nomenclature and atom numbering of tocopherols and chromanols as recommended by IUPAC was used throughout [66, 67]. 1H and 13C NMR resonances of the isoprenoid side chain of tocopherols are only insignificantly influenced (Δ < 0.05 ppm) by modifications of the chroman ring [68, 69], and are thus listed here only once: δ = 19.7 (C-4a′), 19.8 (C-8a′), 21.2 (C-2′), 22.7 (C-13′), 22.8 (C-12a′), 24.6 (C-6′), 24.8 (C-10′), 28.0 (C-12′), 32.6 (C-8′), 32.8 (C-4′), 37.3 (C-7′), 37.4 (C-9′), 37.5 (C-5′), 37.5 (C-3′), 39.3 (C-11′), 39.9 (C-1′) ppm. In the spiro-dimeric compounds 6 and 10, the aromatic moiety is conventionally numbered, while the spiro-keto moiety is indicated by “#”.
para -α-Tocopherylquinone (5)
Dark yellow oil; UV (toluene): λmax = 264 nm; 1H NMR: δ = 2.55 (2H, m, 4-CH2), 2.08 (3H, s, 5a-CH3), 2.03 (3H, s, 7a-CH3), 2.03 (3H, s, 8b-CH3), 1.08–1.54 (m, 23H, 3-CH2, 9 × CH2 in side chain [1′-CH2, 2′-CH2, 3′-CH2, 5′-CH2, 6′-CH2, 7′-CH2, 9′-CH2, 10′-CH2, 11′-CH2], 3 × CH in side chain [4-CH, 8-CH, 12-CH]), 1.23 (3H, s, 2a-CH3), 0.86 (9H, d, J = 6.3 Hz, 4a-CH3, 8a-CH3, 12a-CH3), 0.84 (3H, d, J = 6.3 Hz, 13-CH3) ppm; 13: δ = 187.5 (C-8a), 140.3 (C-8), 12.3 (C-8b), 140.0 (C-7), 12.3 (C-7a), 186.6 (C-6), 140.6 (C-5), 12.0 (C-5a), 144.4 (C-4a), 21.5 (C-4), 40.4 (C-3), 72.7 (C-2), 26.7 (C-2a) ppm; isoprenoid side chain: see above. NMR data fully agree with the literature [25, 70].
Spiro -dimer of α-tocopherol (6)
Yellow oil; UV (toluene): λmax = 272 nm; 1H NMR: δ = 2.62 (2H, m, 4-CH2), 2.46 (2H, m, 4#-CH2), 2.52 (3H, s, 5a-CH2), 2.08 (3H, s, 5a#-CH2), 2.12 (3H, s, 7a-CH3), 1.88 (3H, s, 7a#-CH3), 2.05 (3H, s, 8b-CH3), 2.01 (3H, s, 8b#-CH3), 1.64–1.60 (4H, m, 3-CH2 and 3#-CH2), 1.06–1.54 (m, 42H, 18 × CH2 and 6 × CH in two isoprenoid side chains), 1.22 (3H, s, 2a-CH3), 1.20 (3H, s, 2a#-CH3), 0.82–0.90 (24H, m, 4a-CH3, 8a-CH3, 12a-CH3, 13-CH3, 4a#-CH3, 8a#-CH3, 12a#-CH3, 13#-CH3) ppm; 13C NMR: δ = 143.7 (C-8a), 141.8 (C-8a#), 122.4 (C-8, C-8#), 10.7 (C-8b), 13.0 (C-8b#), 121.1 (C-7), 126.0 (C-7#), 10.8 (C-7a), 10.2 (C-7a#), 144.7 (C-6), 201.4 (C-6#), 114.5 (C-5), 80.0 (C-5#), 16.9 (C-5a), 27.1 (C-5a), 114.0 (C-4a), 114.4 (C-4a#), 18.6 (C-4), 16.6 (C-4#), 29.7 (C-3), 30.2 (C-3#), 73.3 (C-2), 75.0 (C-2), 22.3 (C-2a), 22.5 (C-2a#) ppm; isoprenoid side chains: see above. NMR data are in complete agreement with the literature [26].
α-Tocopheryl- ortho -quinone (α-tocored, 7)
Red oil; UV (toluene): λmax = 288 nm; 1H NMR: δ = 2.44 (2H, m, 4-CH2), 2.05 (3H, s, 7a-CH3), 1.98 (3H, s, 8b-CH3), 1.73 (2H, t, 3-CH2), 1.08–1.54 (m, 21H, 9 × CH2 in side chain [1′-CH2, 2′-CH2, 3′-CH2, 5′-CH2, 6′-CH2, 7′-CH2, 9′-CH2, 10′-CH2, 11′-CH2], 3 × CH in side chain [4-CH, 8-CH, 12-CH]), 1.28 (3H, s, 2a-CH3), 0.84–0.86 (12H, d, J = 6.3 Hz, 4a-CH3, 8a-CH3, 12a-CH3), 0.84 (3H, d, J = 6.3 Hz, 13-CH3) ppm; 13C NMR: δ = 163.5 (C-8a), 110.3 (C-8), 11.7 (C-8b), 134.4 (C-7), 13.8 (C-7a), 180.1 (C-6), 181.0 (C-5), 143.8 (C-4a), 22.6 (C-4), 40.0 (C-3), 71.4 (C-2), 28.0 (C-2a) ppm; isoprenoid side chain: see above. NMR data are fully consistent with the literature [71].
5a-(1-Imidazolyl)-α-tocopherol (8)
1H NMR: δ = 2.11 (3H, s, 7a-CH3), 2.15 (3H, s, 8b-CH3), 2.60 (2H, t, 4-CH2), 3.85 (2H, s, 5a-CH2), 6.83 (s, 1H, H(Im)-5); 7.02 (s, 1H, H(Im)-4), 7.40 (s, 1H, H(Im)-2) ppm; 13C NMR: δ = 11.7 (C-8b), 11.9 (C-7a), 19.4 (C-4), 31.1 (C-3), 51.4 (C-5a), 74.5 (C-2), 114.2 (C-4a), 115.7 (C-5), 124.0 (C-7), 125.3 (C-8), 128.1 (C(Im)-4), 129.3 (C(Im)-5), 138.4 (C(Im)-2), 144.5 (C-8a), 145.7 (C-6) ppm.
Spiro -dimer of N -methyl-α-tocopheramine (10)
Yellow oil; UV (toluene): λmax = 278 nm; 1H NMR: δ = 2.60 (2H, m, 4-CH2), 2.46 (2H, m, 4#-CH2), 2.36 (3H, s, 5a-CH3), 2.18 (3H, s, 5a#-CH3), 2.05 (3H, s, 7a-CH3), 1.84 (3H, s, 7a#-CH3), 2.01 (3H, s, 8b-CH3), 1.96 (3H, s, 8b#-CH3), 1.64–1.59 (4H, m, 3-CH2 and 3#-CH2), 1.06–1.54 (m, 42H, 18 × CH2 and 6 × CH in two isoprenoid side chains), 1.22 (3H, s, 2a-CH3), 1.21 (3H, s, 2a#-CH3), 0.82–0.92 (24H, m, 4a-CH3, 8a-CH3, 12a-CH3, 13-CH3, 4a#-CH3, 8a#-CH3, 12a#-CH3, 13#-CH3) ppm; 13C NMR: δ = 145.4 (C-8a), 142.4 (C-8a#), 122.8 (C-8), 122.6 (C-8#), 10.7 (C-8b), 12.9 (C-8b#), 123.2 (C-7), 125.8 (C-7#), 10.9 (C-7a), 10.4 (C-7a#), 138.6 (C-6), 251.3 (C-6#), 111.2 (C-5), 68.7 (C-5#), 18.8 (C-5a), 25.5 (C-5a), 116.2 (C-4a), 116.0 (C-4a#), 18.8 (C-4), 16.6 (C-4#), 30.2 (C-3, C-3#), 73.8 (C-2), 75.1 (C-2), 22.3 (C-2a, C-2a#), 38.8 (CH3-N), 42.4 (CH3-N =) ppm; isoprenoid side chains: see above.
Ellagic acid (11)
Yellow powder; 1H NMR: δ = 7.46 (H-Ar), 8.12 (OH) ppm; 13C NMR: δ = 107.4 (C-2), 110.2 (C-6) 112.4 (C-1), 136.3 (C-3), 140.1 (C-4), 148.1 (C-5), 159.1 (COO) ppm.
Ellagobis( ortho -quinone) (2,3,5,7,8,10-Hexahydrochromeno[5,4,3- cde ]chromene-2,3,5,7,8,10-hexaone, 12)
Black powder; 1H NMR: δ = 8.05 (CH) ppm; 13C NMR: δ = 116.1 (C-6), 115.9 (C-1), 118.8 (C-2), 145.0 (C-3), 155.9 (COO), 177.5 (C-4), 180.4 (C-5) ppm.
Gallic imidazolide (13)
Yellow needles; 1H NMR: δ = 6.32 (s, 2H, 2-CH, 6-CH), 6.81 (s, 1H, H(Im)-5), 7.08 (s, 1H, H(Im)-4), 7.44 (s, 1H, H(Im)-2) ppm; 13C NMR: δ = 107.9 (C-2, C-6), 125.5 (C-1), 127.2 (C(Im)-4), 129.2 (C(Im)-5), 145.6 (C-3, C-5), 135.5 (C-4), 138.2 (C(Im)-2), 166.1 (CON) ppm.
Hydroxytyrosol ortho -quinone (14)
Red oil; 1H NMR: δ = 2.69 (2H, t, Ar-CH2), 3.70 (2H, t, CH2-OH), 6.60 (1H, dd, H-6), 6.72 (1H, dd, H-2), 6.78 (1H, dd, H-5) ppm; 13C NMR: δ = 31.2 (Ar-CH2) 61.4 (CH2-OH), 127.7 (CH-5), 129.2 (CH-2), 143.2 (CH-6), 151.1 (C-1), 180.3 (C-4), 182.4 (C-3) ppm.
Dihydroxytyrosol (15)
Yellowish wax; 1H NMR: δ = 3.42 (2H, dd, J = 3.6, 8.0 Hz, Ar–CH(OH)), 4.04 (1H, dd, J = 3.6, 8.0 Hz, CH2-OH), 6.54 (1H, dd, J = 2.0, 8.0 Hz, H-6), 6.67 (1H, d, J = 8.0 Hz, H-5), 6.75 (1H, d, J = 2.0 Hz, H-2) ppm; 13C NMR: δ = 66.5 (CH2-OH), 84.9 (Ar–CH(OH)), 115.4 (CH-2), 116.1 (CH-5), 120.3 (CH-6), 131.1 (C-1), 143.6 (C-4), 144.9 (C-3) ppm.
Dihydroxytyrosol ortho -quinone (16)
Red oil; 1H NMR: δ = 3.44 (2H, dd, J = 3.6, 7.9 Hz, Ar–CH(OH)), 3.99 (1H, dd, J = 3.6, 7.9 Hz, CH2-OH), 6.56 (1H, dd, H-6), 6.70 (1H, dd, H-2), 6.76 (1H, dd, H-5) ppm; 13C NMR: δ = 66.1 (CH2-OH), 85.2 (Ar–CH(OH)), 127.2 (CH-5), 128.8 (CH-2), 143.4 (CH-6), 151.9 (C-1), 182.4 (C-4), 185.5 (C-3) ppm.
5,6-Dihydroxybenzofuran (17)
Yellow wax; 1H NMR: δ = 5.23 (2H, br, OH), 6.42 (1H, t, J = 3.2 Hz, H-2), 7.24 (1H, t, J = 3.2 Hz, H-3), 7.42 (1H, d, J = 0.6 Hz, H-4), 7.48 (1H, d, J = 0.6 Hz, H-7) ppm; 13C NMR: δ = 106.1 (CH-7), 106.9 (CH-3), 124.6 (CH-4), 127.9 (C-3a), 143.2 (CH-2), 144.2 (C-5), 150.1 (C-6), 155.4 (C-8) ppm.
5,6-Dihydroxybenzofuran ortho -quinone (18)
Red oil; 1H NMR: δ = 6.33 (1H, t, J = 3.2 Hz, H-2), 6.88 (1H, t, J = 3.2 Hz, H-3), 7.02 (1H, s, H-4), 7.08 (1H, s, H-7) ppm; 13C NMR: δ = 110.1 (CH-3), 126.5 (CH-3a), 131.2 (C-4), 133.4 (CH-7), 145.1 (C-2), 150.1 (C-8), 182.3, 182.6 (C-5, C-6) ppm.
Data availability
Complete analytical data are provided in the article.
References
Sixta H, Iakovlev M, Testova L, Roselli A, Hummel M, Borrega M, van Heiningen A, Froschauer C, Schottenberger H (2013) Cellulose 20:1547
Sixta H, Michud A, Hauru L, Asaadi S, Ma Y, King AWT, Kilpeläinen I, Hummel M (2015) Nord Pulp Paper Res J 30:43
Stepan AM, Michud A, Hellstén S, Hummel M, Sixta H (2016) Ind Eng Chem Res 55:8225
Asaadi S, Kakko T, King AWT, Kilpeläinen I, Hummel M, Sixta H (2018) ACS Sust Chem Eng 6:9418
Guizani C, Larkiala S, Moriam K, Sawada D, Elsayed S, Rantasalo S, Hummel M, Sixta H (2021) J Appl Polym Sci 138:49787
Zhang J, Kitayama H, Potthast A, Rosenau T, Gotoh Y (2019) Carbohydr Polym 226:115258
Zhang J, Yamagishi N, Gotoh Y, Potthast A, Rosenau T (2020) J Appl Polym Sci 137:48681
Ebner G, Schiehser S, Potthast A, Rosenau T (2008) Tetrahedron Lett 49:7322
Zweckmair T, Hettegger H, Abushamalla H, Bacher M, Potthast A, Laborie MP, Rosenau T (2015) Cellulose 22:3583
Hettegger H, Zhang J, Koide M, Rinner U, Potthast A, Gotoh Y, Rosenau T (2022) Fibers 10:50
Stolze K, Udilova N, Rosenau T, Hofinger A, Nohl H (2003) Biol Chem 384:457
Potthast A, Rosenau T, Henniges U, Schiehser S, Kosma P, Saake B, Lebioda S, Radosta S, Vorwerg W, Wetzel H, Koschella A, Heinze T, Strobin G, Sixta H, Strlic M, Isogai A (2015) Cellulose 22:1591
Korntner P, Hosoya T, Dietz T, Eibinger K, Reiter H, Spitzbart M, Röder T, Borgards A, Kreiner W, Mahler AK, Winter H, French AD, Henniges U, Potthast A, Rosenau T (2015) Cellulose 22:1053
Preedy VR, Watson RR (eds) (2007) Encyclopedia of Vitamin E. CABI Publishing, Oxford, Cambridge
Henniges U, Vejdovszky P, Siller M, Jeong MJ, Rosenau T, Potthast A (2014) Curr Chromatogr 1:52
Barbini S, Jaxel J, Karlström K, Rosenau T, Potthast A (2021) Bioresour Technol 341:125862
Barbini S, Sriranganadane D, España Orozco S, Kabrelian A, Karlström K, Rosenau T, Potthast A (2021) ACS Sust Chem Eng 9:1323
Catala A (2012) Tocopherol: sources, uses and health benefits. Nova Science, UK
Dahe GJ, Teotia RS, Kadam SS, Bellare JR (2011) Biomaterials 32:352
Wu HL, Bremner DH, Li HY, Shi QQ, Wu JZ, Xiao RQ, Zhu LM (2016) Mat Sci Eng C 62:702
Ghaheh FS, Khoddami A, Alihosseini F, Jing S, Ribeiro A, Cavaco-Paulo A, Silva C (2017) Process Biochem 59:46
Yokota S, Kitaoka T, Opietnik M, Rosenau T, Wariishi H (2008) Angew Chem Int Ed 47:9866
Rüegg R, Mayer H, Schudel P, Schwieter U, Tamm R, Isler R (1967) Veröffentl Deut Ges Ernährung 16:22
Machlin LJ (1980) Vitamin E: a comprehensive treatise. Marcel Dekker, New York
Schudel P, Mayer H, Metzger J, Rüegg R, Isler O (1963) Helv Chim Acta 46:636
Schröder H, Netscher T (2001) Magn Reson Chem 39:701
Rosenau T, Gruner M, Habicher WD (1997) Tetrahedron 53:3571
Rosenau T, Potthast A, Elder T, Kosma P (2002) Org Lett 4:4285
Rosenau T, Böhmdorfer S (2009) ortho-Quinone methides in tocopherol chemistry. In: Rokita S (ed), Wiley series on reactive intermediates in chemistry and biology, vol 1. Wiley, New York, p 163
Rosenau T, Ebner G, Stanger A, Perl S, Nuri L (2005) Chem Eur J 11:280
Liebner F, Ebner G, Becker E, Potthast A, Rosenau T (2010) Holzforschung 64:161
Rosenau T, Habicher WD (1995) Tetrahedron 51:7919
Smith LI, Renfrow WB, Opie JW (1942) J Am Chem Soc 64:1082
Mayer H, Isler O (1971) Tocopheramines and tocopherthiols. In: Colowick SP, Kaplan NO (eds) Methods in enzymology, vol 18, part C. Academic Press, New York, London, p 275, 334
Blomstrand R, Forsgren L (1968) Int J Vit Res 38:328
Schlegel W, Schwieter U, Tamm R (1969) Non-toxic antioxidants, based on chromane derivatives. US Patent 3458637, Jul 29, 1969; (1968). Chem Abstr69:21909
Søndergaard E, Dam H (1970) Zeitschr Ernährungswiss 10:71
Tokuwame M (1991) Synergistic antioxidant-heat stabilizer systems for polyolefins. JP Patent 03043458, Feb 25. Chem Abstr 115:73015
Rosenau T, Potthast A, Milacher W, Adorjan I, Hofinger A, Kosma P (2005) Cellulose 12:197
Tomic-Vatic A, Eytina J, Chapman J, Mahdavian E, Neuzil J, Salvatore BA (2005) Int J Cancer 117:188
Gille L, Stamberg W, Patel A, Böhmdorfer S, Rosenau T (2012) Free Rad Biol Med 53:100
Zingg JM (2007) Mini Rev Med Chem 7:545
Neuzil J, Tomasetti M, Zhao Y, Dong LF, Birringer M, Wang XF, Low P, Wu K, Salvatore BA, Ralph SJ (2007) Mol Pharmacol 71:1185
Tanito M, Yoshida Y, Kaidzu S, Chen ZH, Cynshi O, Jishage KI, Niki E, Ohira A (2007) Investig Ophthalm Visual Sci 48:396
Bieri JG, Mason KE (1968) J Nutr 96:192
Rosenau T, Hofinger A, Potthast A, Kosma P (2004) Org Lett 6:541
Patel A, Rosenau T (2021) Monatsh Chem 152:1231
Patel A, Liebner F, Netscher T, Mereiter K, Rosenau T (2007) J Org Chem 72:6504
Adegoke GO, Vijay Kumar M, Gopala Krishna AG, Varadaraj MC, Sambaiah K, Lokesh BR (1998) J Food Sci Technol 35:283
Ding T, Li T, Li J (2019) ACS Appl Mat Interfaces 11:6463
Wendler F, Meister F, Heinze T (2005) Macromol Symp 223:213
Britton J, Davis R, O’Connor KE (2019) Appl Microbiol Biotechnol 103:5957
Quiles JL, Ramírez-Tortosa MC, Yaqoob P (eds) (2006) Olive oil and health. CABI Publishing, Wallingford
Peltzer M, Navarro R, López J, Jiménez A (2010) Polym Degrad Stab 95:1636
Fortunati E, Luzi F, Fanali C, Dugo L, Belluomo GM, Torre L, Kenny JM, Santi L, Bernini R (2017) J Renew Mat 5:81
Haddar W, Baaka N, Meksi N, Ticha MB, Guesmi A, Mhenni MF (2015) Fibers Polym 16:1506
Bayraktar O (2018) Textile Leather Rev 1:90
Elder T, Del Río JC, Ralph J, Rencoret J, Kim H, Beckham GT, Crowley MF (2020) ACS Sustain Chem Eng 8:11033
Yamauchi F, Ito T, Kawamoto O, Komatsu T, Akiyama T, Yokoyama T, Matsumoto Y (2020) Holzforschung 74:559
Miyagawa Y, Tobimatsu Y, Lam PY, Mizukami T, Sakurai S, Kamitakahara H, Takano T (2020) Plant J 104:156
Zaccaron S, Ahn K, Henniges U, Potthast A, Rosenau T (2022) Cellulose 29:3733
Hosoya T, Bacher M, Potthast A, Elder T, Rosenau T (2018) Cellulose 25:3797
ISO 2470-1 (2009) Measurement of diffuse blue reflectance factor—Part 1: indoor daylight conditions (ISO brightness). International Organization for Standardization, Geneva
Brightness loss of pulp. TAPPI method UM 200 (2012) TAPPI, Peachtree Corners, GA
Standard Testing Methods. Paptac E.4P (2012) Pulp and Paper Technical Association of Canada. Brossard (Quebec), Canada
IUPAC-IUB Commission on Biochemical Nomenclature (CBN) (1982) Eur J Biochem 123:473
IUPAC-IUB Commission on Biochemical Nomenclature (CBN) (1974) Arch Biochim Biophys 165:1
Urano S, Hattori Y, Yamanoi S, Matsuo M (1980) Chem Pharm Bull 28:1992
Brownstein S, Ingold KU (1989) J Org Chem 54:560
Urones JG, Marcos IS, Cubillo L, Garrido NM, Basabe P (1990) Phytochem 29:2223
Yamauchi R, Matsui T, Kato K, Ueno Y (1990) Agric Biol Chem 54:2703
Acknowledgements
The financial support of the Austrian Biorefinery Center Tulln (ABCT-II) is gratefully acknowledged. The county of Lower Austria is gratefully acknowledged for the support through the project WST3-F-5030820 (Stabilization of polymers through antioxidants).
Funding
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosenau, T., Potthast, A., Zhang, J. et al. Cellulosic fibers spun from stabilized imidazolium ionic liquids: identification of chromophores in the fibers after accelerated ageing. Monatsh Chem (2024). https://doi.org/10.1007/s00706-024-03203-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00706-024-03203-6