
Abstract
Capillary electrophoresis methods for the separation of carbohydrates with four different detection techniques, namely direct UV, indirect UV, capacitively coupled conductivity, and laser-induced fluorescence detection, were tested and their performance was evaluated and compared in terms of linearity, limits of detection and quantitation, repeatability, recovery, analysis time, and sample treatment. The test set of analytes comprised sucrose, glucose, and fructose. The effect of using lactose as an internal standard on the individual methods was investigated, too. The results showed that laser-induced fluorescence detection is a technique of choice for applications requiring the detection of very low amounts of reducing carbohydrates. Contactless conductivity detection is favorable when detection sensitivity is not a crucial parameter but fast and reliable analysis is required. When only a UV detector is available as a standard part of capillary electrophoresis instruments, direct UV detection can be used when analysis time is not a critical parameter. For fast analysis with UV detectors, indirect UV detection is the technique of choice. Finally, to verify the applicability of the tested methods, samples of cola beverage, honey, and orange juice were analyzed and the results obtained by all four methods were compared.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Saccharides have an important role in various biological processes, such as the source and storage of energy, cell signaling, and structural support. They significantly influence biological activity, the conformation of molecules, and the metabolism of glycoproteins and glycolipids. Due to their diverse roles in biological systems, there is an increased interest in their analysis in many fields, including food science, medicine, and biochemistry [1,2,3]. Analytical techniques used for identifying and quantifying saccharides are thus crucial in various applications. These techniques include high-performance liquid chromatography (HPLC), gas chromatography (GC), paper chromatography, and thin-layer chromatography (TLC) [4,5,6,7]. Saccharides are problematic analytes and each method has its advantages and disadvantages, the choice of appropriate technique thus depends on various factors such as the matrix of the sample, required sensitivity, and available instrumentation. Different modes of HPLC can be used, such as reversed-phase HPLC, hydrophilic interaction liquid chromatography, or high-performance anion-exchange chromatography with pulsed amperometric detection [8]. When using GC for saccharide analysis, the analytes have to be derivatized prior to their analysis due to their lack of volatility. With that, the risk of contamination of the sample or its loss is higher. GC is often coupled with mass spectrometry or flame ionization detector [9]. Paper chromatography and TLC are inexpensive and simple methods for the quick separation of saccharides based on their different affinity to the stationary and mobile phases [6, 7]. Capillary electrophoresis (CE) proved to be a well-suited technique for separating and determining saccharides, too. In the case of CE, commonly used detection techniques include UV detection, electrochemical detection, fluorescence detection, and mass spectrometry (MS) detection.
UV detection
Direct UV detection of saccharides without derivatization is not typically used due to the lack of chromophore in the molecule causing low absorbance in the UV range. One possibility to overcome this issue is to use highly alkaline buffers with a pH higher than the pKa of carbohydrate hydroxyl groups (pH > 12). In the strongly basic environment, hydroxyl groups of carbohydrates are dissociated and form enediolates containing a semi-aromatic system that can be detected at 270 nm [10]. Another method uses the complexation with borate buffer and detection at 195 nm, nevertheless, this detection method exhibits low selectivity as many other compounds absorb at this wavelength [11].
During indirect UV detection, a UV probe that absorbs UV radiation is added to the background electrolyte (BGE). The probe must be of the same charge as the analyte because the detection is based on the displacement of the probe by the analyte in the analyte zone due to the electroneutrality conservation law. When the zone of the analyte migrates through the detector the absorbance drops, showing a negative peak in the electropherogram. The wavelength is chosen based on the additive, such as 214 nm (1,2,4-tricarboxybenzoic acid or 5-sulfosalicylic acid) [12], 215 nm, and 207 nm (glycylglycin) [13], 254 nm (sorbic acid) [14], or 400 nm (p-nitrophenol) [15].
Electrochemical detection
Detection based on electrochemical principles is divided into 3 types, amperometric, conductometric, and potentiometric detection. Amperometric detection is a highly sensitive method allowing the detection of low concentrations of saccharides, however, the high voltage used for separation can disrupt the detection. The saccharides are oxidized on the working electrode and the current that is generated is detected. Pulsed amperometric detection [16, 17] and amperometric detection at constant potentials [18, 19] were used in the CE analysis of saccharides in several cases.
Conductivity detection depends on the difference in the conductivity of the analyte zone and BGE. It can be performed in contact or contactless mode. Capacitively coupled contactless conductivity detection (C4D) is particularly popular. When using a highly alkaline BGE, the hydroxyl groups of saccharides are dissociated and thus they show a negative charge and migrate in the electric field. However, their migration is rather slow compared to hydroxide anions present in BGE, and thus their molar conductivity is significantly lower. As a result, the zone of the analyte has lower conductivity than BGE, which is manifested by negative peaks on the C4D signal. However, with increasing pH of the BGE, the conductivity also increases causing a higher current during separation, which can lead to overheating of the solution inside the capillary. To overcome this problem, capillaries with smaller inner diameters can be used because the sensitivity of the C4D detection is not reduced as it would be with UV detection [20,21,22].
Laser-induced fluorescence detection (LIF)
LIF can be used after labeling saccharides with an appropriate fluorescence tag. This method is highly sensitive and very low concentrations of saccharides can be detected. On the other hand, the derivatization process is time-consuming and laborious. It also increases the risk of sample loss or contamination during the analysis process. Derivatization agents commonly used for labeling saccharides are for example 8-aminopyrene-1,3,6-trisulfonic acid (APTS), 7-amino-1,3-naphthalenedisulfonic acid, and 4-amino-5-hydroxynaphthalene-2,7-disulfonic acid [23,24,25].
MS detection
Lastly, MS detection is used for identifying and quantifying a wide range of saccharides. They are ionized in an electrospray interface and analyzed based on their mass-to-charge ratio. To combine CE with MS detection for saccharide analysis, using volatile BGEs with high pH is necessary, for example, diethylamine solution is a suitable BGE [26, 27]. Nevertheless, the CE-MS instrumentation does not belong to common equipment. The interfacing of CE and MS is significantly more troublesome than the liquid chromatography-MS connection, and compared to the other mentioned detection techniques, MS is more demanding in terms of acquisition and operational costs, as well as operator qualification and experience.
In this work, we have compared the performance and some practical aspects of four detection techniques, direct and indirect UV, C4D, and LIF, in the detection of saccharides, using glucose, fructose, and sucrose as model analytes with lactose as an internal standard. The analytes and the internal standard were chosen based the on previous experience of our group [20].
Results and discussion
To evaluate and compare the performance of the individual detection techniques, calibration dependences were measured and evaluated with and without using lactose as an internal standard. From these dependences, determination coefficients were calculated. The limit of detection (LOD) and limit of quantitation (LOQ) were determined as analyte concentrations providing peak heights corresponding to the three- and ten-fold baseline noise, respectively. Then, standard carbohydrate mixtures at two different concentration levels depending on the calibration range were prepared and measured ten times. From these measurements, the RSD values of absolute and relative peak areas and migration times were calculated. Recovery was expressed as the carbohydrate concentration found in these samples related to the real concentration. The aforementioned performance parameters were then compared and some specific features of individual detection techniques and methods were considered, too. Detailed figures of merit for all three analytes and four methods tested can be found in Supplementary Material (Table S1-S4). A concise overview of the results for comparison of the individual methods is provided in Table 1.
Direct UV detection
Direct UV detection was evaluated using a method published by Rovio et al. [10]. This method uses a high-pH BGE, 36 mmol dm−3 Na2HPO4 with 130 mmol dm−3 NaOH, pH = 12.6, which apart from the dissociation of carbohydrate hydroxyl groups leads to the formation of semi-aromatic enediolate absorbing at 270 nm. The high pH of the BGE is connected with its high conductivity, resulting in high currents and excessive Joule heating. Suppressing the heating by using capillaries of smaller inner diameters dramatically lowers the sensitivity of UV detection, and thus classical 50-µm i.d. capillary is used and the overheating is avoided by applying lower voltage (16 kV) with a 2-min potential ramp at the beginning of the separation, and decreased temperature (17 °C). This results in a time-consuming separation, in which the analytes pass through the detector after 20–35 min (Fig. 1A). We tested the possibility of shortening the analysis time by applying low pressure (1.0 kPa) to the capillary inlet during the separation. This led to a significant reduction of migration times of the analytes to 17–23 min (Fig. 1B). On the other hand, the introduction of the pressure-driven flow with a parabolic profile caused significant peak tailing with glucose and fructose peaks beginning to overlap. Original conditions without the additional pressure were thus conserved. Overall, it should be noted that the method is highly sensitive to even seemingly insignificant alterations of the experimental protocol, e.g., flushing the capillary with 10% acetic acid before each run, long potential ramp, etc. Calibration standards were prepared in the concentration range from 50 to 500 µg cm−3 with lactose added as an internal standard at the concentration of 500 µg cm−3. Higher concentrations than 500 µg cm−3 were not included because the calibration curve deviated from linearity. The repeatability and recovery were tested using samples at 70 and 400 µg cm−3 concentration levels.

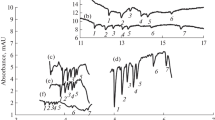
Separation of carbohydrates with direct UV detection at 270 nm. Identification of analytes: 1. sucrose, 2. lactose, 3. glucose, 4. fructose. Concentration 500 µg cm−3. Fused-silica capillary 50 µm i.d., 375 µm o.d., 65.0 cm total and 56.5 cm effective length. BGE 36 mM Na2HPO4 with 130 mM NaOH, pH = 12.6. Separation voltage 16 kV (2-min potential ramp) without (A) and with (B) additional pressure of 1.0 kPa applied to the capillary inlet, current 112 µA. Temperature 17 °C. Sample injection 5.0 kPa for 4 s followed by 5.0 kPa for 5 s injection of BGE
As can be seen in Table S1, the method showed rather low determination coefficients when absolute peak area values were used to construct calibration curves. Relating the peak areas to lactose improved the linearity and all three R2 values were above 0.99. The regression model thus explained 99% of the data variability. The migration time repeatability was also significantly improved by relating the migration times to lactose with RSDs below 1% for the absolute and RSDs below 0.4% for the relative migration times. Relating the peak areas to the internal standard had only a small effect on RSDs at the lower concentration level with all RSDs below 4% but it significantly improved peak area repeatability at the higher level with RSDs up to 5.25% reduced to 2.50%. For this method, the usage of the internal standard thus brought a significant improvement. The method showed good recovery at both concentration levels (89.5–108.5). LOD values ranged from 1.0 to 1.7 µg cm−3, and LOQ was between 5.2 and 8.8 µg cm−3.
Indirect UV detection
Indirect UV detection was evaluated using a method based on Gürel et al. [13], which was slightly modified. A 50 mmol dm−3 glycylglycine adjusted to pH = 12.3 with NaOH was used as BGE. In this BGE, high pH ensured dissociation and electromigration of carbohydrates while glycylglycine, present in its anionic form served as a UV probe for indirect detection at 207 nm. A 50-µm i.d. capillary was used to keep the sensitivity of UV detection as high as possible. The original method worked with 28 kV voltage, nevertheless, the excessive Joule heating under these conditions led to an unstable signal baseline. To stabilize the signal, we have lowered the voltage to 20 kV. Separation was completed in 5.5 min with all four peaks baseline separated (Fig. 2). It should be noted that even with lowered voltage, the baseline was rather unstable, which was a source of certain inaccuracy in data evaluation. Calibration standards were prepared in the concentration range from 100 to 1 000 µg cm−3 with lactose added as an internal standard at the concentration of 500 µg cm−3. The repeatability and recovery were tested using samples at 200 and 700 µg cm−3 concentration levels.

Separation of carbohydrates with indirect UV detection at 207 nm. Identification of analytes: 1. sucrose, 2. lactose, 3. glucose, 4. fructose. Concentration 1 000 µg cm−3, lactose 500 µg cm−3. Fused-silica capillary 50 µm i.d., 375 µm o.d., 50.0 cm total and 41.5 cm effective length. BGE 50 mmol dm−3 glycylglycine, adjusted with NaOH to pH = 12.3. Separation voltage 20 kV, current 63 µA. Temperature 25 °C. Sample injection 5.0 kPa for 5 s
Table S2 summarizes the method performance parameters evaluated. Determination coefficients for calibrations with absolute peak areas were in two of three cases higher than 0.999 showing excellent linearity. Using relative peak areas somewhat lowered the determination coefficients but they remained higher than 0.99. Using lactose as an internal standard increased the RSDs of peak areas for glucose and fructose and slightly decreased the RSD for sucrose. RSD for absolute peak areas was below 5% for all analytes at both concentration levels. For relative peak areas, RSDs increased up to 8.83%. Only for migration times, using the internal standard significantly increased the repeatability. RSDs were reduced from the 0.46–0.80% range for absolute values to 0.04–0.32% for the relative ones. The overall effect of using lactose as an internal standard can thus be considered negative. The recovery for the higher concentration level was very good, ranging from 98.6 to 105.7%. For the lower concentration, the recovery values were probably negatively influenced by baseline instability as they ranged from 75.0 to 125.0%. These problems were also reflected by higher LOD and LOQ values in the range of 32–46 µg cm−3 and 95–154 µg cm−3, respectively.
C4D detection
To evaluate C4D, a method used in our previous work was chosen [20]. The advantage of conductivity detection is that, unlike UV or LIF detection, its sensitivity is not dramatically reduced by using capillaries with smaller inner diameters. On the contrary, the higher separation efficiency brought by smaller inner diameter capillaries leads to increased sensitivity. We consider 20-µm i.d. capillary as a good compromise because the lower current values in narrow-bore capillaries go hand in hand with more efficient heat dissipation but they have a higher tendency to clog and higher pressure/time is needed for sample injection and conditioning of the capillaries. A 40 mmol dm−3 NaOH, pH = 12.5, was used as BGE ensuring the dissociation of carbohydrates. High resolution of analyte peaks under the standard setup allowed for reducing the separation time by using the short-end injection approach. The 15 kV separation voltage, inducing a current of only 10 µA, resulted in a stable baseline signal. Separation was completed within 3 min (Fig. 3). The analytes, having significantly lower electrophoretic mobility than BGE hydroxide anions, exhibited negative peaks on the conductivity signal. Calibration standards were prepared in the concentration range from 5 to 500 µg cm−3 with lactose added as an internal standard at the concentration of 100 µg cm−3. Higher concentrations than 500 µg cm−3 were not measured as the peaks of glucose and fructose began to overlap. The repeatability and recovery were tested using samples at 20 and 200 µg cm−3 concentration levels.

Separation of carbohydrates with C4D detection. Identification of analytes: 1. sucrose, 2. lactose, 3. glucose, 4. fructose. Concentration 200 µg cm−3, lactose 100 µg cm−3. Fused-silica capillary 20 µm i.d., 375 µm o.d., 50.0 cm total and 15.0 cm effective length. BGE 40 mmol dm−3 NaOH, pH = 12.5. Separation voltage 15 kV, current 10 µA. Temperature 25 °C. Sample injection 5.0 kPa for 20 s
Table S3 summarizes the performance parameters evaluated for the C4D method. Determination coefficients for all three calibrations with absolute peak areas and two of three calibrations with relative peak areas were higher than 0.999, the last one was 0.9989. The method thus exhibited excellent linearity. Although, using relative peak areas slightly reduced the determination coefficients. RSDs of absolute peak areas were excellent at the higher concentration level (0.51–0.60%) and increased but remained below 2% for relative peak areas. RSDs at the lower level were below 3.5% for glucose and fructose, but up to 10% for sucrose. Using the internal standard here resulted in a very slight increase in RSDs. The effect of using the internal standard on migration time repeatability was ambiguous, nevertheless, all migration time RSDs were 1.01% or lower. Overall, the effect of internal standard was neither significantly positive nor negative. As the calibration range of this method was extended over two orders of magnitude, the recovery for the lower concentration level was between 85.3 and 122.1%, which was rather unsatisfactory. Recovery in the lower concentration range was improved by using weighted linear regression. Weighting coefficients of 1/x, 1/x2, and 1/x3 were tested with the 1/x2 coefficient exhibiting the best values of recovery and determination coefficients. As can be seen in Table S3, weighted calibration curves showed slightly lower determination coefficients because the weighted linear regression prefers a better fit of the lower concentration points that have a weaker effect on the R2 value than the higher concentration points. Recovery at both concentration levels ranged from 92.3 to 107.7. LOD and LOQ values were 1.0–1.7 µg cm−3 and 3.3–5.6 µg cm−3, respectively.
LIF detection
To assess the performance of LIF detection, a method based on the conditions reported by Monsarrat et al. [28] and Yang et al. [29] was used. APTS was used as the fluorescent label. Apart from fluorescence the APTS label also provided the analytes with three negative charges from three sulfonate groups. Thanks to that, an acidic BGE consisting of 30 mmol dm−3 TEA and 1 mol dm−3 acetic acid, pH = 3.1 could be used. Due to the lower conductivity of this BGE, − 30 kV voltage could be used in a 50-µm i.d. capillary with a current of only − 25 µA. In acidic BGE, the electroosmotic flow was suppressed, and negative polarity of voltage was used for the separation of quickly migrating triply negatively charged APTS derivatives. As a result, the separation was completed in 3 min (Fig. 4). A disadvantage of reductive amination labeling by APTS is that only reducing carbohydrates can be derivatized. Furthermore, fructose cannot be efficiently labeled by this procedure, too [30]. From the test set of analytes, only glucose and lactose could thus be labeled. Calibration standards were prepared in the concentration range from 0.01 to 0.70 µg cm−3 of glucose with lactose added as an internal standard at the concentration of 0.70 µg cm−3. The repeatability and recovery were tested using samples at 0.04 and 0.30 µg cm−3 concentration levels.

Separation of carbohydrates with LIF detection. Identification of analytes: 1. APTS-glucose, 2. APTS-lactose. Concentration of glucose 0.3 µg cm−3, lactose 0.7 µg cm−3. Fused-silica capillary 50 µm i.d., 375 µm o.d., 50.0 cm total and 29.0 cm effective length. BGE 30 mmol dm−3 TEA and 1 mol dm−3 acetic acid, pH = 3.1. Separation voltage − 30 kV, current − 25 µA. Temperature 25 °C. Sample injection 5.0 kPa for 5 s
The method showed excellent linearity with R2 higher than 0.999 (Table S4). The repeatability of the peak area was significantly improved by relating peak areas to lactose at both concentration levels as RSD values were reduced from up to 8.59% to no higher than 5%. The repeatability of migration time at the higher concentration level was also improved by relating to lactose. The RSD decreased from 1.61 to 0.52%. Using lactose as an internal standard thus has a positive effect on the method’s performance. Recovery was 91.1% and 88.0% at the lower and the higher concentration level, respectively. Compared to the other detection techniques, the LOD and LOQ were extremely low, 2.1 and 6.4 ng cm−3, respectively.
Comparison of the detection techniques
Table 1 provides a concise overview of the figures of merit of all four tested methods. When comparing the individual test parameters, C4D and LIF detection showed excellent linearity, manifested by very high determination coefficients. The lowest R2 values were, on the contrary, obtained for direct UV detection. Concerning the LOD and LOQ values, LIF superseded the other techniques by 3–4 orders of magnitude in the case of glucose. For sucrose and fructose that could not be APTS-labeled the lowest LOD and LOQ values were achieved using direct C4D. Direct UV detection LOD and LOQ were only negligibly higher. On the other hand, LOD and LOQ of indirect UV detection were more than one order of magnitude higher.
Concerning repeatability, migration time RSDs were below 2% in all cases and below 1% in most cases. No straightforward conclusion about the repeatability of peak areas can be made. Very low RSDs (below 2%) were observed at higher concentration levels for C4D, and LIF with internal standard. Most peak area RSD values were below or around 5%. Most of the methods showed good recovery (within the range of 90–110%). Recoveries lying significantly out of this range were obtained with indirect UV detection at the lower concentration level, which can be ascribed to the lower baseline stability. In the case of C4D at the lower concentration level, the recoveries were improved by using the weighted linear regression of calibration curves. Recovery slightly below 90% was observed for LIF, which can be caused by the complicated sample pre-treatment.
Using lactose as an internal standard significantly improved the performance of the direct UV and LIF methods. It did not bring significant improvement to the C4D method and deteriorated the performance of the indirect UV method. It should be added that, in our experience, relating the peak areas to an internal standard can help to maintain method reliability in long-term usage. The short-term testing performed in this study did not allow us to evaluate this effect.
Concerning other parameters, due to their specific features, C4D and LIF detection techniques allow for very fast separation finished within 3 min. On the contrary, direct UV detection requires very specific conditions that lead to very long migration times.
When considering LIF detection, the laborious and time-consuming sample pretreatment connected with the consumption of the expensive labeling agent should be considered, too. The other three methods do not require any sample pretreatment.
To verify the applicability and performance of the tested methods in the analysis of real samples, three different samples were analyzed, i.e., cola beverage, honey, and orange juice from concentrate. Figure 5 shows an overview of the determined concentration or content of individual carbohydrates. In the samples of cola beverage and honey, only glucose and fructose were detected. The content of sucrose was in both cases below LOD. The application of the method with LIF detection to the analysis of the orange juice sample was unsuccessful. Injection of this sample resulted in distorted peaks and prolonged migration times. The peak shape was further deteriorating during repeated injections. Flushing the capillary with 1 M NaOH did not resolve this problem. Thus, the glucose content in orange juice could not be determined by the LIF method. Concentrations of glucose and fructose found in cola and honey samples were mostly in good agreement. In some cases, the concentration determined by the indirect UV detection method was slightly higher which can be ascribed to the lower sensitivity of the method, leading to less precise determination. Glucose content in honey determined by the LIF detection method was also slightly higher. This can be caused by very high dilution of the sample. The results obtained for the orange juice sample by direct and indirect UV detection methods are in relatively good agreement. The concentration of glucose and fructose determined by the C4D method is higher. The reason is unclear, and it is probably connected with the rather problematic matrix of this sample.

Content of carbohydrates determined in three samples: A cola beverage, B honey, and C orange juice from concentrate. Results were obtained by the four tested methods with different detection techniques–direct UV (white bars), indirect UV (light gray bars), C.4D (dark gray bars), and LIF (black bars). Median values are plotted with error bars showing the standard deviation (n = 3, α = 0.05)
Conclusion
To conclude, the choice of the detection technique for CE separation of carbohydrates should be always done after careful consideration of the aim of the analysis. From the detection techniques tested, LIF should be always considered when very low detection limits are required. However, the fact that not all carbohydrates can be labeled should be kept in mind.
Sensitivity is often not the limiting factor as many real-life samples for the determination of carbohydrates are rather concentrated and need to be diluted. In such cases, other detection techniques, not requiring sophisticated and expensive special equipment, with no need for laborious sample treatment can be used. C4D and direct UV detection exhibit similar performance considering repeatability, LOD, LOQ, and recovery. The disadvantage of direct UV detection is a very long separation time so when C4D is available it should be considered a better option. Indirect UV detection of carbohydrates shows lower sensitivity and worse recovery at lower concentration levels. It can be considered when only a UV detector is available and analysis time is an important parameter.
Analysis of cola beverage, honey, and orange juice samples showed that the tested methods provide similar results. The results obtained by indirect UV detection can be influenced by lower baseline stability. The accuracy of the results obtained by LIF detection can be affected by the extremely large dilution necessary for successful analysis. From the three matrices tested, orange juice was found to be problematic as it made determination of glucose by the LIF method impossible and probably also negatively influenced the accuracy of the C4D method.
Experimental
Glycylglycin (≥ 99%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). D-Glucose monohydrate, p.a., D-fructose p.a., D-lactose monohydrate p.a., and sodium hydroxide, p.a. were purchased from Penta (Prague, Czech Republic). Sucrose p.a. was supplied by Lachema (Neratovice, Czech Republic). Samples and BGEs were prepared using deionized water produced by the Milli-Q system from Millipore (Danvers, USA). For pH measurements, Jenway 3540 pH/conductivity meter from Jenway (Staffordshire, UK) was used. The dry block heating system QBD2 used for the derivatization of saccharides was purchased from Grant Instruments (Shepreth, UK). A fused-silica capillary was purchased from Polymicro Technologies (Phoenix, USA). An Agilent 7100 CE instrument from Agilent Technologies (Waldbronn, Germany) equipped with a diode array detector was used for all electrophoretic measurements. The CE system was further equipped with a C4D detector from Admet (Prague, Czech Republic), consisting of two cylindrical electrodes, 4 mm long with a 1-mm insulation gap. The inner diameter of the electrodes was 400 µm. The detector was operated at a frequency of 1.84 MHz with an amplitude of 44 V. ZetaLIF LED-induced fluorescence detector from Adelis (Labege, France) with 480 nm excitation wavelength was used for CE-LIF experiments.
Samples of cola beverage, honey, and orange juice from concentrate were purchased in a local grocery store. The orange juice sample was filtered through a 0.20 µm PVDF filter. The cola beverage sample was degassed in an ultrasonic bath for 10 min and then filtered through a 0.20 µm PVDF filter. From the honey sample, 3.6 mg were weighed into a 10-cm3 volumetric flask and filled up with deionized water. Then, the sample was filtered through a 0.20 µm PVDF filter. The samples were eventually further diluted according to the sensitivity of the tested methods.
Direct UV
Unmodified fused-silica capillary, 50 µm ID, 375 µm OD, 65.0 cm total, 56.5 cm effective length was used. Between consecutive runs, the capillary was rinsed for 3 min with 10% v/v acetic acid and 2 min with BGE (disodium phosphate, 36 mmol dm−3, and sodium hydroxide, 130 mmol dm−3 at pH = 12.6). Samples were injected into the capillary using a pressure of 5 kPa for 4 s with consecutive injection of BGE using the same pressure for 5 s. A separation voltage of 16 kV was applied (linearly increasing from 0 kV within the first 2 min), inducing a current of approximately 112 µA. The temperature of the capillary cassette was maintained at 17 °C. The UV detector was set at 270 nm wavelength. Samples of cola beverage and orange juice were diluted 100 × with deionized water prior to analysis. The honey solution was injected without further dilution.
Indirect UV
Unmodified fused-silica capillary, 50 µm ID, 375 µm OD, 50.0 cm total, 41.5 cm effective length was used. Between consecutive runs, the capillary was rinsed for 4 min with BGE (glycylglycine, 50 mmol dm−3, adjusted to pH = 12.3 using NaOH). Samples were injected into the capillary using a pressure of 5 kPa for 3 s. A separation voltage of 20 kV was applied, inducing a current of approximately 63 µA. The temperature of the capillary cassette was maintained at 25 °C. The UV detector was set at 207 nm wavelength. Samples of cola beverage and orange juice were diluted 100 × with deionized water prior to analysis. The honey solution was injected without further dilution.
C4D
Unmodified fused-silica capillary, 20 µm ID, 375 µm OD, 50.0 cm total, 15.0 cm effective length was used. Between consecutive runs, the capillary was rinsed for 4 min with BGE (sodium hydroxide, 40 mmol dm−3). Samples were injected into the capillary using a pressure of 5 kPa for 20 s. A separation voltage of 15 kV was applied, inducing a current of approximately 10 µA. The temperature of the capillary cassette was maintained at 25 °C. Samples of cola beverage and orange juice were diluted 1000 × with deionized water prior to analysis. The honey solution was diluted 2.5 × with deionized water.
LIF
Unmodified fused-silica capillary, 50 µm ID, 375 µm OD, 50.0 cm total, 29.0 cm effective length was used. Between consecutive runs, the capillary was rinsed for 2 min with BGE (triethylamine, 30 mmol dm−3, acetic acid, 1 mol dm−3, pH = 3.1). Samples were injected into the capillary using a pressure of 5 kPa for 5 s. A separation voltage of − 30 kV was applied, inducing a current of approximately 25 µA. The temperature of the capillary cassette was maintained at 25 °C. The cola and orange juice samples were diluted 1000 × with deionized water. 1 mm−3 of the diluted sample was put together with 1 mm−3 of 100 mmol dm−3 APTS in 15% (v/v) acetic acid. Then, 5 mm−3 of 1 mol dm−3 sodium cyanoborohydride was added and the mixture was kept at 70 °C for 1 h. The derivatized samples were further diluted 100 × prior to analysis, resulting in a total dilution factor of 1/700,000. From the honey sample solution, 1 mm−3 was taken for the same derivatization procedure as in the case of the other samples. Following the derivatization, this sample was diluted 250 × with deionized water.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information file. Should any raw data files be needed they are available from the corresponding author upon reasonable request.
References
Solá RJ, Griebenow K (2009) J Pharm Sci 98:45
Oefner P, Chiesa C (1994) Glycobiol 4:397
Paulus A, Klockow A (1996) J Chromatogr A 720:353
Bernal JL, Del Nozal MJ, Toribio L, Del Alamo M (1996) J Agric Food Chem 44:507
Lehotay SJ, Hajšlová J (2002) Trends Anal Chem 21:686
Bourne EJ, Lees EM, Weigel H (1963) J Chromatogr A 11:253
Ovodov YS, Evtushenko E, Vaskovsky VE, Ovodova R, Solov’eva T (1967) J Chromatogr A 26:111
Morales V, Corzo N, Sanz ML (2008) Food Chem 107:922
Ruiz-Matute AI, Hernández-Hernández O, Rodríguez-Sánchez S, Sanz ML, Martínez-Castro I (2011) J Chromatogr B 879:1226
Rovio S, Yli-Kauhaluoma J, Sirén H (2007) Electrophoresis 28:3129
Hoffstetter-Kuhn S, Paulus A, Gassmann E, Widmer HM (1991) Anal Chem 63:1541
Damn J, Overklift G (1994) J Chromatogr A 678:151
Gürel A, Hızal J, Öztekin N, Erim FB (2006) Chromatographia 64:321
Jager AV, Tonin FG, Tavares MFM (2007) J Sep Sci 30:586
Plocek J, Chmelik J (1997) Electrophoresis 18:1148
O’Shea TJ, Lunte SM, LaCourse WR (1993) Anal Chem 65:948
LaCourse WR, Owens GS (1996) Electrophoresis 17:310
Voegel PD, Zhou W, Baldwin RP (1997) Anal Chem 69:951
Ye J, Baldwin RP (1994) J Chromatogr A 687:141
Křížek T, Müllerová R (2020) Monatsh Chem 151:1231
Tůma P, Opekar F, Dlouhý P (2022) Food Chem 375:131858
Tomnikova A, Kozlík P, Křížek T (2022) Electrophoresis 43:1963
Wolff MW, Bazin HG, Lindhardt RJ (1999) Biotechnol Tech 13:797
Starr CM, Masada RI, Hague C, Skop E, Klock JC (1996) J Chromatogr A 720:295
Čokrtová K, Mareš V, Křížek T (2023) Electrophoresis 44:35
Klampfl CW, Buchberger W (2001) Electrophoresis 22:2737
Simó C, Barbas C, Cifuentes A (2005) Electrophoresis 26:1306
Monsarrat B, Brando T, Condouret P, Nigou J, Puzo G (1999) Glycobiology 9:335
Yang B, Mai TD, Tran NT, Taverna M (2022) J Sep Sci 45:3594
van der Burg D, Josefsson L, Mikkonen S, Chotteau V, Emmer A, Wätzig H, Sänger-van de Griend C (2021) Electrophoresis 43 922
Acknowledgements
The work was supported in part by Charles University, project SVV260690, and by the Central European Exchange Program for University Studies, network RO-0010-17-2223 – Teaching and Learning Bioanalysis. SA was supported through IRES US National Science Foundation award #1658615. Any opinions, findings, and conclusions, or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSF.
Funding
Open access publishing supported by the National Technical Library in Prague.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vlčková, N., Šimonová, A., Ďuriš, M. et al. Detection techniques for carbohydrates in capillary electrophoresis – a comparative study. Monatsh Chem 154, 967–975 (2023). https://doi.org/10.1007/s00706-023-03109-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-023-03109-9