
Abstract
Using high-throughput sequencing, we identified a novel carlavirus sequence in a 28-year-old ‘Kotsifali’ grapevine sample collected in Heraklion (Crete, Greece). Using RT-PCR and 5'/3' RACE together with Sanger sequencing, the complete genome sequence of 8299 nt was confirmed and found to contain five open reading frames (ORFs) but to lack an ORF6, which is present in some members of the genus Carlavirus. The novel sequence is most similar to those of two carlaviruses infecting caper, and taking into account the ICTV nomenclature, we propose the name "grapevine carlavirus 1" for this new virus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Betaflexiviridae is a large family of viruses that infect a broad range of plants. It comprises 15 different genera, including the genus Carlavirus. Carlaviruses are non-enveloped, filamentous viruses that vary in length between 600 and 700 nm and are 12–15 nm in diameter. Virions contain a single molecule of positive-sense, single-stranded RNA, 7.4 to 8.7 kb in length, containing a 5' cap and 3' polyA tail. The genome typically contains six open reading frames (ORFs), with ORF1 encoding a 215–225 kDa protein functioning as the viral replicase. ORF 2, 3, and 4 encode the conserved triple gene block (TGB) proteins, while ORF5 encodes a coat protein of 32–36 kDa, and ORF6 encodes a cysteine-rich protein (11–16 kDa) with RNA binding activity. All carlaviruses are transmitted mechanically, while several are also known to be transmitted by insect vectors [1].
Grapevine (Vitis sp.) is host to the largest number of viruses among the cultivated plant species [2]. At least 86 viruses have been identified in grapevine, including 17 different Betaflexiviridae members. These include some economically important viruses such as grapevine virus A, grapevine virus B, and grapevine rupestris stem pitting-associated virus. However, none of the known Betaflexiviridae members infecting grapevine belong to the genus Carlavirus. In this article, we describe the identification and characterization of a novel carlavirus identified in grapevine in Crete.
In 2019 and 2020, ~ 400 grapevine samples were collected from the four prefectures (Chania, Rethymnon, Heraklion, and Lasithi) of the island of Crete, Greece. Selected samples, mostly from grapevines older than 10 years, were pooled and sequenced using high-throughput sequencing (HTS). One of these pools comprised five samples of the local grape cultivars Vilana (two plants), Kotsifali (two plants), and Mandilari (one plant), all grafted onto 140 Ruggeri rootstock, from a single location in the prefecture of Heraklion. The Vilana grapevines were 36 years old, while the Kotsifali plants were 28 years old, and the Mandilari plant was 10 years old.
For sequencing, total RNA was extracted from leaves using a Spectrum™ Plant Total RNA Kit (Sigma-Aldrich), including an on-column DNase I (Roche) digestion, and 1 µg of RNA from each sample was pooled for HTS. Library construction was performed using a TruSeq® Stranded Total RNA with Ribo-Zero™ Plant Kit (Illumina) following the manufacturer’s protocol, and 44,461,174 paired-end reads of 101 bp were generated. Read quality was assessed using FastQC [3], quality and adapter trimming were performed using fastp [4], and ribosomal RNA sequences were removed using BBDuk [5] with the default settings and the reference file (ribokmers.fa) included in the BBTools package. Host-derived sequences were removed by alignment with the genome sequence of V. vinifera (GenBank no. GCA_000003745.2) using BBSplit [5], applying the default settings. The remaining reads were then assembled de novo using SPAdes [6] with the -meta option. Assembled contigs larger than 200 nt were subjected to a BLASTn search (task -blastn, e-value 1E-5) using NCBI BLAST + v. 2.9.0 [7] against the Reference Viral Database (RVDB) v20.0 [8]. Taxonomic information about the BLAST hits of interest was added using the taxonomizr package [9].
BLAST analysis showed that 43 contigs had a high degree of similarity to plant virus or viroid sequences, including those of several well-characterized viruses, like grapevine leafroll-associated viruses 1, 4, and 6, grapevine Pinot Gris virus, and grapevine Roditis leaf discoloration-associated virus, as well as the viroids grapevine yellow speckle viroid 1 and hop stunt viroid (Supplementary Table S1). One contig of 8282 nt appeared to represent the nearly complete genome sequence of a novel Betaflexiviridae member, with only 70.5% nt sequence identity to the carlavirus caper latent virus (CapLV). To confirm the presence of the novel sequence in the pooled samples and to determine the full-length genome sequence, a series of primers were designed (Supplementary Table S2) and used for RT-PCR. cDNA was generated using 500 ng of total RNA and random primers using M-MuLV reverse transcriptase (Minotech, Greece) according to the manufacturer’s instructions. PCR was then carried out using Taq DNA polymerase (Minotech, Greece). Initially, the primer pair Carla-D-F/Carla-D-R (Supplementary Table S2) was used to determine which sample(s) within the pool contained the target sequence. Sample number 3 from cultivar Kotsifali produced the expected PCR product of 860 nt, while no amplification was observed when the other four samples were tested (Fig. 1A). The amplicon was excised and sequenced by the Sanger method (Genewiz, Germany), and the sequence of the amplified fragment was 99% identical to the HTS contig. Next, PCR and sequencing of the complete viral genome were carried out using the remaining primer sets (Supplementary Table S2). PCR with each primer pair generated an amplicon of the expected size, which was sequenced as described above (Supplementary Fig. S1A). To confirm the sequences of the 5' and 3' ends of the viral genome, 5' and 3' RACE were carried out using a SuperScript™ III One-Step RT-PCR System (Invitrogen, USA) according to the manufacturer’s protocol, and amplified fragments were sequenced.

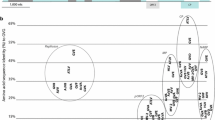
Identification and characterization of a carlavirus from grapevine in Crete. (A) PCR screening of the five pooled grapevine samples to identify the source plant of the novel carlavirus sequence (using primers for part D, see Supplementary Table S2). (B) Schematic representation of the complete genome of grapevine carlavirus 1. Bars correspond to the overlapping PCR fragments that were amplified and sequenced by the Sanger method to verify the HTS results (results in Supplementary Fig. S1). (C and D) Phylogenetic trees based on amino acid sequences of the CP and replicase proteins, using MUSCLE and the maximum-likelihood method with 1000 bootstrap replicates
The complete genome sequence of the novel virus was compiled from the overlapping PCR products and RACE sequences and comprised 8299 nt, excluding the polyA tail (GenBank no. OQ363854). Comparison of the original HTS contig and the Sanger sequence revealed they were almost identical (99.94%). Putative ORFs were identified using ORF Finder [10], with the complete genome predicted to encode five ORFs. ORF1 encodes a polyprotein of 228 kDa containing motifs for methyltransferase, helicase, and RNA-dependent RNA polymerase (RdRp) activity. ORFs 2, 3, and 4 encode the TGB proteins of 31, 12, and 7.2 kDa, respectively, while ORF5 was predicted to encode a CP of 34.8 kDa. No ORF6 was identified based on this analysis (Fig. 1B).
The nt sequence of the complete genome was compared with published sequences, using BLAST, with the grapevine carlavirus sequence having approximately 72% nt sequence identity to soybean carlavirus 1 and jasmine virus C, as well as approximately 71% nt sequence identity to several other carlaviruses, including CapLV (Supplementary Table S3). Based on the current ICTV guidelines for carlaviruses, a member of a new virus species should possess less than 80% amino acid (aa) sequence identity to all known viruses in its polymerase or CP coding gene (https://ictv.global/report_9th/RNApos/Betaflexiviridae), but concerns about these demarcation criteria have been raised recently [11]. Using the replicase aa sequence, the grapevine carlavirus showed 79% identity to caper carlavirus 1 and 78% identity to CapLV, suggesting that it is a novel member of the genus. Interestingly, the CP aa sequence identity was higher than the proposed threshold, with 87.5% aa sequence identity to caper carlavirus 1 and 86.6% identity to CapLV. Finally, the grapevine carlavirus sequence had less than 65% aa sequence identity to other members of the genus (Supplementary Table S3). Phylogenetic analysis was performed using ModelFinder and IQ-Tree [12, 13] following alignment with MUSCLE. The trees were generated using the maximum-likelihood method, using the LG + F + I + G4 model as the best fit for polymerase and the LG + F + G4 model for CP, with 1000 bootstrap replicates [14], and visualized using TreeView [15]. The replicase and CP sequences of the grapevine carlavirus isolate clustered together with the two caper carlaviruses (Fig. 1C and D), confirming that these are the most closely related viruses in the genus.
Based on the host range and replicase and CP aa sequence identity values, the virus reported in this paper is distinct from other known carlaviruses. Accordingly, we have named it "grapevine carlavirus 1". The complete genome has a typical organization for members of the genus Carlavirus although it lacks the small ORF6 found in many members of the genus. Interestingly, genome sequence alignments with the two closest caper-infecting viruses identified an insertion of 266 nt in the TGB genes (nt 6939–7205) in grapevine carlavirus 1, suggesting significant genome variation despite the high degree of similarity in the CP coding region. Importantly, no symptoms were observed in the specific vine sample from which the sequence was obtained (Supplementary Fig. S1B). Further work to identify other viruses in this accession will be useful in case symptoms are observed at another time point during the growing season, as well as for testing the mechanical transmissibility of the virus, which is typical of the genus.
References
Adams MJ, Candress T, Hammond J et al (2012) Betaflexiviridae. Virus Taxonomy. Elsevier Inc., pp 920–941
Fuchs M (2020) Grapevine viruses: a multitude of diverse species with simple but overall poorly adopted management solutions in the vineyard. J Plant Pathol 102:643–653. https://doi.org/10.1007/s42161-020-00579-2
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890. https://doi.org/10.1093/bioinformatics/bty560
Bushnell B BBmap - https://sourceforge.net/projects/bbmap/
Nurk S, Bankevich A, Antipov D et al (2013) Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. https://doi.org/10.1089/cmb.2013.0084
Camacho C, Coulouris G, Avagyan V et al (2009) BLAST+: Architecture and applications. BMC Bioinformatics 10:1–9. https://doi.org/10.1186/1471-2105-10-421
Goodacre N, Aljanahi A, Nandakumar S et al (2018) A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 3:1–18. https://doi.org/10.1128/mspheredirect.00069-18
Sherrill-Mix S (2021) taxonomizr: Functions to Work with NCBI Accessions and Taxonomy. Available at: https://cran.r-project.org/web/packages/taxonomizr/index.html
NCBI ORF Finder. Available at https://www.ncbi.nlm.nih.gov/orffinder/
Silva JMF, Melo FL, Elena SF et al (2022) Virus classification based on in-depth sequence analyses and development of demarcation criteria using the Betaflexiviridae as a case study. J Gen Virol 103:0–16. https://doi.org/10.1099/jgv.0.001806
Tamura K, Stecher G, Kumar S (2021) MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Hoang DT, Chernomor O, von Haeseler A et al (2018) UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol Biol Evol 35:518–522. https://doi.org/10.1093/molbev/msx281
Jaime HC, Serra F, Bork P (2016) ETE3: Recontruction, analysi and viualization of phylogenomic data. Mol Biol Evol 33(6):1635–1638. https://doi.org/10.1093/molbev/msw046
Acknowledgements
The authors would like to thank the National Flagship Initiative “The paths of Grapevine” of the public investments programs of the GSRT:2018ΣE0100000. Funding for this project came from a category C grant entitled ‘The effect of changes in Rural communities may affect the plant-pathogen interactions’ KA10894-Special Account for Research Funds of University of Crete.
Funding
Open access funding provided by HEAL-Link Greece.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Statement and declaration
The authors have no relevant financial or non-financial interests to disclose, and no bioethical issues to address.
Additional information
Communicated by Sead Sabanadzovic
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material
Additional file 1: Supplementary Fig. S1
(A) Overlapping PCRs for characterization and sequencing of the complete genome. (B) Photo of the Kotsifali grapevine from which leaf tissue was sampled
Additional file 2: Supplementary Table S1
List of viruses identified by HTS
Additional file 3: Supplementary Table S2
Primers used to characterize the novel grapevine carlavirus
Additional file 4: Supplementary Table S3
BLAST results of the complete nucleotide (nt) or polymerase and coat protein amino acid (aa) sequences of the novel grapevine virus against both assigned and unassigned members of the genus Carlavirus
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Katsarou, K., Andronis, C., James, A. et al. Complete genome sequence of a carlavirus identified in grapevine (Vitis sp) in Greece. Arch Virol 168, 172 (2023). https://doi.org/10.1007/s00705-023-05795-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05795-6