
Abstract
Hand, foot, and mouth disease (HFMD) is a common childhood infection caused by human enteroviruses and is clinically characterised by fever with vesicular rash on the hands, feet, and mouth. While enterovirus A71 (EV-A71) and coxsackievirus A16 (CVA16) were the major etiological agents of HFMD in India earlier, the data on recently circulating enteroviruses associated with HFMD are sparse. Here, we describe the molecular epidemiology of enteroviruses associated with HFMD in South India from 2015 to 2017. We used archived enterovirus real-time reverse transcription (RT) PCR-positive vesicle swab and/or throat swab specimens from clinically suspected HFMD cases collected from four secondary-care hospitals in South India between July 2015 and December 2017. PCR amplification and sequencing were done based on the 5’VP1, 3’VP1, VP2, or 5´NCR regions to identify enterovirus types. Genetic diversity among enteroviruses was inferred by phylogenetic analysis. Of the 107 enterovirus RNA real-time RT-PCR-positive HFMD cases, 69 (64%) were typed as CVA6, 16 (15%) were CVA16, and one (1%) was CVA10, whereas in 21 (20%) cases, the virus was not typeable by any of the methods used in the study. The majority of HFMD cases (89, 83%) were in children less than five years old, while 11 (10.3%) were in adults. 5’VP1 yielded the maximum number of enteroviruses genotyped, and phylogenetic analysis showed that the CVA6 strains belonged to subclade D3, while the subclades of CVA16 and CVA10 were B1c and D, respectively. The predominant etiological agent of HFMD in South India during 2015-2017 was CVA6, followed by CVA16 and CVA10.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hand, foot, and mouth disease (HFMD) is an acute febrile infection caused by enteroviruses (EVs) that is mainly found in children less than five years of age and is clinically characterised by fever and typical vesicular lesions on the hands, feet, mouth, or buttocks [1]. Enteroviruses are single-stranded RNA viruses belonging to the genus Enterovirus, family Picornaviridae [2]. Although HFMD is a self-limiting infectious disease, it can also lead to severe clinical manifestations such as meningitis, encephalitis, and myocarditis in children [3,4,5]. Since the 1950s, HFMD has been reported in the form of outbreaks and sporadic cases, caused mainly by coxsackievirus A16 (CVA16), enterovirus 71 (EV-A71), coxsackievirus A6 (CVA6), coxsackievirus A10 (CVA10), and other EVs of the EV-A, EV-B, and EV-C groups [6, 7]. The transmission of HFMD occurs mainly through direct contact with mucus, saliva, fluid from blisters, or feces of an infected patient and indirectly by fomites [8].
India reported the first outbreak of HFMD in 2003, in children from Kerala with mild symptoms, and this outbreak was caused by EV-71 [9]. In the following years, significant morbidity and mortality were reported in children with EV-71 infection in Uttar Pradesh from 2004 to 2006 [10]. In 2007, an epidemic of HFMD was reported in children with mild symptoms from West Bengal [11]. Since then, other enterovirus types, such as CVA16, CVA6, EV-A71, CVA10, and E-9, have been circulating sporadically in India, causing mild-to-severe infections in children [12,13,14,15,16]. Recent reports from Europe, North America, and Asia have indicated the emergence of CVA6-associated HFMD outbreaks and sporadic cases, while CVA16, EV-A71, and CVA10 have been sparsely distributed [17, 18]. CVA6 is one of the major enterovirus strains identified in both mild and severe cases of HFMD globally in recent years [19, 20].
While there have been insufficient data on the etiology and clinico-epidemiological profile of HFMD cases from South India in recent years, our retrospective study mainly focused on the molecular epidemiology and clinical features of HFMD in South India from July 2015 to December 2017.
Materials and methods
Clinical specimens and collection of epidemiological data
Archived pan-enterovirus real-time RT-PCR-positive vesicle swab and/or throat swab specimens of clinically diagnosed HFMD [1] cases collected as a part of the sentinel surveillance in 2015 and those received for virological diagnosis in 2016 and 2017 were used in this study. Any clinically diagnosed HFMD cases with vesicular lesions anywhere in the body with or without fever and from South India during the period of July 2015 to December 2017 were enrolled. The specimens were anonymised using a unique identifier. Clinical and epidemiological data were obtained from the case record forms or laboratory request forms.
Enterovirus detection and typing
The algorithm for enterovirus detection and typing is shown in Figure 1. Total RNA was extracted from vesicle and throat swab specimens using a QIAamp Viral RNA Mini Kit (QIAGEN, Germany) as per the manufacturer’s protocol. The extracted RNA was subjected to pan-enterovirus real-time reverse transcription (RT) PCR using a Respiratory Pathogens 21 Kit (Fast Track Diagnostics - FTD, Luxembourg). When vesicle and throat swabs were available, vesicle swabs were screened first, and if negative, only the throat swabs were tested.

The algorithm of enterovirus detection and typing in clinical specimens
Multi-locus-specific PCR and sequencing enabled the typing of enteroviruses associated with HFMD. Enteroviruses from the positive samples were typed by PCR based on portions of the 5’VP1, 3’VP1, VP2, and 5’NCR regions, followed by Sanger sequencing. Pan-enterovirus nested RT-PCR was performed by initial cDNA synthesis and subsequent nested RT-PCR targeting the 5’VP1 region (PCR-1, ~992 bp; PCR-2, ~375 bp) [21]. If PCR based on the 5’VP1 region failed, VP2-based semi-nested RT-PCR was performed (PCR-1, ~583 bp; PCR-2, ~367 bp) [22]. Subsequently, nested RT-PCR targeting the 5’NCR region was carried out (PCR-1, ~529 bp; PCR-2, ~397 bp) when other regions failed to be amplified [23]. CVA6-specific semi-nested RT-PCR was carried out for samples collected in 2015 based on the 3’VP1 region (PCR-1, ~657 bp; PCR-2, ~420 bp) [24]. For each PCR, the reaction mix contained forward and reverse primers (Supplementary Table S1), AgPath-ID One-Step RT-PCR reagents (Applied Biosystems, USA), and the extracted RNA. All PCR assays were performed by previously described methods [21,22,23,24]. A BigDye Terminator V3.1 Cycle Sequencing Kit was used to sequence purified PCR products in an ABI-3500 Genetic Analyser (Applied Biosystems, USA). The sequence quality was analysed and sequences were edited using Sequencher 5.4.6 software (Gene Codes Corporation, USA). BLAST was used to determine the percentage of sequence identity to reference strains in the GenBank database (NCBI). Viruses that could not be typed by the above methods were subjected to CVA16- and EV-A71-specific real-time RT-PCR targeting the VP1 locus as described elsewhere [25, 26].
Phylogenetic analysis
All of the study sequences were aligned with reference sequences obtained from GenBank- NCBI by multiple sequence alignment in ClustalW (Supplementary Table S2). Phylogenetic analysis was performed using the software MEGA X (version 10.2.5) [27]. Phylogenetic dendrograms based on partial sequences of the 5’VP1, 3’VP1, VP2, and 5’NCR regions were constructed using the neighbour-joining method with the Kimura 2-parameter (K2P) model. Bootstrap values (≥70%) were determined for each node, using 1000 replicates to assess the statistical reliability of the tree. Phylogenetic trees were constructed based on four different target regions (5’VP1, 3’VP1, VP2, and 5’NCR), but only the sequences that were amplified from the corresponding targets were included in the trees, and no samples were duplicated.
Statistical analysis of the demographic and clinical data was done using STATA/SE software (version 14.1).
Results
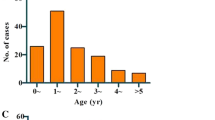
A total of 172 archived samples from clinically suspected HFMD cases were included in this study. Of these, 148 samples were collected from secondary-care hospitals in Kozhikode, 18 were from Wayanad, four were from Malappuram district of Kerala state, and two were from the Shimoga district of Karnataka state in South India. Of these 172 samples, 43 (25.0%) were collected in 2015, 65 (37.8%) in 2016, and 64 (37.2%) in 2017. Enterovirus RNA was detected in 107 (62%) out of 172 clinically suspected HFMD cases by pan-enterovirus real-time RT-PCR. Of the suspected HFMD cases tested, enterovirus RNA was detected in 27 (25.2%), 40 (37.4%), and 40 (37.4%) of the samples collected in 2015, 2016, and 2017, respectively. Of the 107 positive cases, 89 (83%) of the individuals were below five years of age, and 18 (17%) were between 5 and 65 years of age (Table 1). Eleven (10.3%) adults exhibited signs of HFMD. The number of affected males was 68 (64%), while 39 (36%) were females. The mean age of the patients with HFMD was 4.4 years, with age ranging from 9 months to 65 years. The mean age of the male and female population was 5.4 and 2.8 years, respectively. The most common clinical feature observed in the enterovirus-associated HFMD cases was vesicular rash with fever (90 out of 107 cases, 84%). Respiratory symptoms such as cough (18, 16%), coryza (6, 6%), breathlessness (1, 1%), and sore throat (9, 8%) were also observed. Some of the HFMD patients had oral ulcers (3, 3%), headache (4, 4%), vomiting (2, 2%), myalgia (1, 1%), abdominal pain (1, 1%), or lymphadenopathy (1, 1%). Examples of rash in patients with hand, foot, and mouth disease are shown in Figure 2. We also observed other rash types, such as papules (25, 23%), maculopapular rash (19, 18%), crusted lesions (11, 10%), macules (7, 5%), erythema (8, 8%), pustules (3, 3%), erosion (3, 3%), and bullae (1, 1%). Rash was seen on the hands (87, 81%), feet (91, 85%), mouth (51, 48%), soles (39, 36%), palms (41, 38%), buttocks (32, 30%), and other parts of the body (<20%).

Rash in children with hand, foot, and mouth disease. (A) erythematous macules on the palms, (B) crusted lesions on the knee, (C) papules on the foot, (D) erythematous macules on the soles
From 2015 to 2017, both vesicle and throat swabs were collected from 46 patients, while only vesicle swabs were collected from 115 patients and only throat swabs were collected from 11 patients. Of the 107 laboratory-confirmed cases of HFMD, enterovirus RNA was detected in vesicular (97, 91%) and throat swabs (10, 9%). Of the 86 (80%) typed enteroviruses, 69 (64%) were CVA6, 16 (15%) were CVA16, and one (1%) was CVA10. However, 21 (20%) of the enteroviruses that were positive in real-time PCR could not be typed by any of the molecular typing strategies used in this study. In 2015, 24 HFMD cases were caused by CVA6, while one was caused by CVA16. In 2016, 20 HFMD cases were caused by CVA6 infection, while nine were caused by CVA16. In the following year, 2017, 25 cases of HFMD were caused by CVA6, six by CVA16, and one by CVA10. HFMD cases were predominantly found in the rainy season from June to November.
The PCR and sequencing enabled CVA6 typing in 27 samples based on 5’VP1, five based on VP2, and nine based on 5’NCR. CVA6-specific nested RT-PCR based on the 3’VP1 region enabled the typing of CVA6-strains in the other 28 samples, but sequencing was possible with only 15 samples. Out of 16 (15%) samples positive for CVA16, the strain was identified by PCR and sequencing in six samples based on 5’VP1, in two samples based on VP2, and one sample based on 5’NCR. CVA16-specific real-time RT-PCR identified CVA16 in the other seven samples. CVA10 was identified in only one sample (1%) by 5’VP1-based PCR and sequencing. However, 21 samples were not typed by any of the enterovirus typing methods used. A total of 66 sequences obtained in this study were deposited in the GenBank database under the accession numbers MG885750, MG885751, MG840455-MG840478, MG840480-MG840487, MG869706-MG869710, MG885752-MG885768, MH160045-MH160049, and MH160051-MH160055.
Phylogenetic dendrograms of CVA6 based on partial 5’VP1 (398 bp), 3’VP1 (475 bp), VP2 (540 bp), and 5’NCR (301 bp) sequences are shown in Figure 3. In the tree based on partial 5’VP1 sequences (n = 74), 22 CVA6 strains clustered together with other Indian strains (MH539787, 2016) with 96-100% nucleotide sequence identity, followed by five strains with 97-97.5% sequence identity to an Australian strain (MH111055, 2017) of subclade D3 (Fig. 3A). The phylogeny based on 3’VP1 sequences (n = 67) showed 14 CVA6 strains clustered together with an Indian strain (MH539787, 2016), with 96.2-98.7% identity, and one with 98.1% sequence identity to an Australian strain (MH111055, 2017) of subclade D3 (Fig. 3B). The phylogeny based on VP2 sequences (n = 39) showed two CVA6 subclade D3 strains with 99.1-99.3% sequence identity to an Indian strain (MH539787, 2016), while the other three strains formed a cluster with an Australian strain (MH111055, 2017), with 96.9-97.78% identity (Fig. 3C). In the tree based on 41 partial 5’NCR sequences (301 bp), four CVA6 strains showed 99.0% identity to an Indian strain (MH539787, 2016), while the other five strains showed 98-99% identity to an Australian strain (MH111055, 2017) of subclade D3 (Fig. 3D). Phylogenetic trees based on the 5’VP1 (413 bp), VP2 (533 bp), and 5’NCR (301 bp) regions are shown in Figure 4. The phylogenic tree based on 5’VP1 (n = 34) showed five CVA16 strains with 96.4–97.6% identity to an Indian strain (KY792583, 2013) and one with 98.3% identity to another Indian strain (KY792578, 2013) of the B1c subclade (Fig. 4A). In the tree based on VP2, two CVA16 strains showed 97.2% nucleotide sequence identity to an Indian strain (KY792584, 2015) (n = 33) (Fig. 4B). One CVA16 strain was 99% identical to Indian strain (KY792577, 2012) based on 5’NCR sequences (n = 42) (Fig. 4C). A CVA10 strain from the study showed 95.6% identity to an Indian strain (MH118041, 2017) of subclade D based on the 5’VP1 region (410 bp, n = 33) (Fig. 5). The inter-clade nucleotide sequence divergence was 13.6-30.9% for 5’VP1, 13.1-24.0% for 3’VP1, 15.6-19.2% for VP2, and 4.7-11.3% for 5’NCR. The inter-clade nucleotide sequence divergence was 24.9-27.9% for 5’VP1, 21.4-24.8% for VP2, and 5.7-10.0% for 5’NCR. The inter-clade nucleotide sequence divergence based on 5’VP1 was 14.4-27.8%.

Phylogenetic trees of CVA6 based on partial 5’VP1 (A), 3’VP1 (B), VP2 (C), and 5’NCR (D) sequences of the study strains and other representative global strains. Phylogenetic relationships were inferred using the neighbour-joining method with the Kimura 2-parameter (K2P) model with 1000 bootstrap replicates for each data set. The GenBank accession number and the place and year of collection are shown for each strain. The clades and subclades are colour coded from node to tip (clade A in dark green, clade B in yellow, clade C in blue, subclades D1 in pink, D2 in light green, and D3 in red). The study strains are indicated by blue text. The scale bar represents nucleotide substitutions per site.

Phylogenetic analysis of CVA16 based on partial 5’VP1 (A), VP2 (B), and 5’NCR (C) sequences of the study strains and reference strains. Phylogenetic relationships were inferred using the neighbour-joining method with the Kimura 2-parameter (K2P) model. The statistical reliability was estimated for each node using 1000 bootstrap replicates. The branches are colour coded as clade A in dark green, subclade B2 in yellow, subclade B3 in blue, subclades B1a in pink, B1b in violet, and B1c in red. The study strains are indicated by blue text. The scale bar represents nucleotide substitutions per site.

Phylogenetic tree of the CVA10 strain from this study and global representative strains, based on partial 5’VP1 sequences. Phylogenetic relationships were inferred using the neighbour-joining method with the Kimura 2-parameter (K2P) model with 1000 bootstrap replicates. The clades and subclades are colour coded from node to tip (clade A in dark green, clade B in yellow, clade C in blue, clade D in red, clade E in violet, F in pink, and G in light green). The strain from this study is indicated by blue text. The scale bar at the bottom indicates the relative phylogenetic distance.
Discussion
From July 2015 to December 2017, the predominant enterovirus type associated with HFMD in South India was CVA6. This is in contrast to earlier reports from 2003 to 2008, where EV-A71 and CVA16 were associated with HFMD both sporadically and in outbreaks [9, 11]. Since then, the cocirculation of CVA16, CVA6, EV-A71, CV-10, and E-9 has been reported to cause sporadic infections in India [12, 13]. Here, we report the cocirculation of CVA6, CVA16, and CVA10 in South India from 2015 to 2017, where CVA6 was the predominant strain, followed by CVA16 and CVA10. CVA6 has emerged recently as the globally predominant etiological agent of HFMD, while other enteroviruses have been sparsely distributed in recent years [5, 17, 18, 28, 29]. It is worth noting the absence of EV-A71 in this study, which indicates low circulation of this strain in South India. HFMD mainly affects children below five years of age, more frequently males than females [30, 31]. Typical HFMD presentation with upper respiratory tract symptoms and atypical rashes such as crusted lesions, macules, erythema, pustules, erosion, and bullae were observed in the patients.
The multi-locus PCR and sequencing approach enabled the typing of 80% of the enteroviruses found in clinical specimens. By targeting the 5’VP1, 3’VP1, VP2, and 5’NCR regions, the enteroviruses were typed as CVA6, CVA16, and CVA10. The highest success rate for enterovirus typing has been achieved by PCR and sequencing of structural-protein-coding genes such as the 5’VP1, 3’VP1, and VP2 [21, 22, 24]. Of the four strategies used in the study, sequencing based on 5’VP1 yielded the largest number of enteroviruses successfully typed. Enteroviruses that could not be typed by the previously attempted methods were sequenced using PCR products based on the 5’NCR. Hence, the 5’NCR can also be a target for the genotyping of enteroviruses, with certain limits of accuracy. Targeting the 5’NCR region for PCR and sequencing will be helpful in genotyping of enteroviruses when amplification of other genomic regions by PCR fails [23]. Unsuccessful genetic identification of enteroviruses in clinical specimens may be due to an insufficiently high viral load, RNA degradation, frequent genetic mutations, or recombination [32]. The nucleotide sequence variability among enteroviruses in the subclades of CVA6, CVA16, and CVA10 was high in the 5’VP1, 3’VP1, and VP2 regions, while the 5’NCR showed less genetic divergence, making 5’VP1, 3’VP1, and VP2 more suitable for studying evolutionary relationships among enterovirus strains than the 5’NCR. Phylogenetic analysis based on different sequence regions showed that most of the CVA6 strains clustered together with other Indian strains, while some of them grouped with viruses sampled in Australia. Our findings indicate the presence of genetically similar CVA6 strains in India and Australia. Phylogenetic evidence shows that this virus might have emerged during large outbreaks in mainland China between 2012 and 2013. Our findings corroborate earlier studies showing the predominant circulation of CVA6 strains of Asian origin in the Asia-Pacific region in recent years [29, 33,34,35]. These finding increases the concern over the emergence and spread of global strains of CVA6. The CVA16 and CVA10 strains detected during the study period were genetically similar to virus strains reported in others parts of India.
The present investigation might have missed mild or subclinical cases that occurred during this period, as this was a hospital-based HFMD surveillance. Further, the retrospective study using archived samples might have introduced bias in sampling and may not fully represent the distribution of the cases in the study period. Enterovirus typing was not possible for 20% of clinical specimens by any of the molecular methods used in the study. Further investigations should be focused on typing and characterisation of unidentified enteroviruses by next-generation sequencing (NGS).
There is insufficient laboratory surveillance data on the etiology of HFMD cases in India. The present study adds new knowledge and contributes to our understanding of circulating enterovirus types associated with HFMD cases in India and their molecular epidemiology. The data from this study may contribute to the development of an evidence-based outbreak preparedness plan and selection of strains for vaccine development. The changing epidemiology of HFMD, including the emergence of new enterovirus types, emphasises the need for systematic laboratory-based HFMD surveillance in disease-endemic regions like India. In conclusion, over the years, CVA6 has emerged as the predominant etiological agent of HFMD in South India.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Pacific WHORO for the W. A guide to clinical management and public health response for hand, foot and mouth disease (HFMD) [Internet]. Manila : WHO Regional Office for the Western Pacific; 2011. https://iris.wpro.who.int/handle/10665.1/5521. Accessed 24 May 2020
Identification and molecular characterization of non-polio enteroviruses from children with acute flaccid paralysis in West Africa, 2013–2014 [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5476622/. Accessed 29 May 2019
Kim B, Moon S, Bae GR, Lee H, Pai H, Oh SH (2018) Factors associated with severe neurologic complications in patients with either hand-foot-mouth disease or herpangina: a nationwide observational study in South Korea, 2009–2014. Lau EHY, editor. PLoS ONE 13(8):e0201726
Qiu J, Yan H, Cheng N, Lu X, Hu X, Liang L et al (2019) The Clinical and Epidemiological Study of Children with Hand, Foot, and Mouth Disease in Hunan, China from 2013 to 2017. Sci Rep 9(1):1–7
Esposito S, Principi N (2018) Hand, foot and mouth disease: current knowledge on clinical manifestations, epidemiology, aetiology and prevention. Eur J Clin Microbiol Infect Dis 37(3):391–398
Robinson CR, Doane FW, Rhodes AJ (1958) Report of an Outbreak of Febrile Illness with Pharyngeal Lesions and Exanthem: Toronto, Summer 1957—Isolation of Group A Coxsackie Virus. Can Med Assoc J 79(8):615–621
Duong V, Mey C, Eloit M, Zhu H, Danet L, Huang Z et al (2016) Molecular epidemiology of human enterovirus 71 at the origin of an epidemic of fatal hand, foot and mouth disease cases in Cambodia. Emerg Microbes Infect. 5(9):e104
Transmission of Hand, Foot and Mouth Disease and Its Potential Driving Factors in Hong Kong | Scientific Reports [Internet]. https://www.nature.com/articles/srep27500. Accessed 8 May 2022
Sasidharan CK, Sugathan P, Agarwal R, Khare S, Lal S, Jayaram Paniker CK (2005) Hand-foot-and-mouth disease in Calicut. Indian J Pediatr 72(1):17–21
Beig FK, Malik A, Rizvi M, Acharya D, Khare S (2010) Etiology and clinico-epidemiological profile of acute viral encephalitis in children of western Uttar Pradesh. India. Int J Infect Dis. 14(2):e141-146
Sarma N, Sarkar A, Mukherjee A, Ghosh A, Dhar S, Malakar R (2009) Epidemic of hand, foot and mouth disease in west Bengal, India in August, 2007: a multicentric study. Indian J Dermatol 54(1):26–30
Gopalkrishna V, Patil PR, Patil GP, Chitambar SD (2012) Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J Med Microbiol 61(3):420–425
Saxena VK, Pawar SD, Qureshi THIH, Surve P, Yadav P, Nabi F et al (2020) Isolation and molecular characterization of coxsackievirus A6 and coxsackievirus A16 from a case of recurrent Hand, Foot and Mouth Disease in Mumbai, Maharashtra, India, 2018. VirusDis. 31(1):56–60
Sabitha S, Sasidharanpillai S, Sanjay R, Binitha M, Riyaz N, Muhammed K, et al (2018) Clinical profile and virology analysis of hand, foot and mouth disease cases from North Kerala, India in 2015–2016: A tertiary care hospital-based cross-sectional study. Indian J Dermatol Venereol Leprol 0(0):0.
Rao CD, Yergolkar P, Shankarappa KS (2012) Antigenic diversity of enteroviruses associated with nonpolio acute flaccid paralysis, India, 2007–2009. Emerg Infect Dis 18(11):1833–1840
Laxmivandana R, Yergolkar P, Gopalkrishna V, Chitambar SD (2013) Characterization of the non-polio enterovirus infections associated with acute flaccid paralysis in South-Western India. PLoS ONE 8(4):e61650
Bian L, Wang Y, Yao X, Mao Q, Xu M, Liang Z (2015) Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther 13(9):1061–1071
Anh NT, Nhu LNT, Van HMT, Hong NTT, Thanh TT, Hang VTT, et al. Emerging Coxsackievirus A6 Causing Hand, Foot and Mouth Disease, Vietnam—volume 24, Number 4—April 2018 - Emerging Infectious Diseases journal - CDC. https://wwwnc.cdc.gov/eid/article/24/4/17-1298_article. Accessed 18 Apr 2020
Yang X, Li Y, Zhang C, Zhan W, Xie J, Hu S et al (2020) Clinical features and phylogenetic analysis of severe hand-foot-and-mouth disease caused by Coxsackievirus A6. Infect Genet Evol 1(77):104054
Aswathyraj S, Sabeena S, Bhat KG, Bharani KC, Sanjay R, Arunkumar G (2018) Coxsackievirus A6 (CV-A6) Encephalomyelitis in an immunocompromised child-a case report and brief review of the literature. Jpn J Infect Dis 71:388–389
Nix WA, Oberste MS, Pallansch MA (2006) Sensitive, Seminested PCR Amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J Clin Microbiol 44(8):2698–2704
Nasri D, Bouslama L, Omar S, Saoudin H, Bourlet T, Aouni M et al (2007) Typing of human enterovirus by partial sequencing of VP2. J Clin Microbiol 45(8):2370–2379
Richter J, Koptides D, Tryfonos C, Christodoulou C (2006) Molecular typing of enteroviruses associated with viral meningitis in Cyprus, 2000–2002. J Med Microbiol 55(Pt 8):1035–1041
Puenpa J, Chieochansin T, Linsuwanon P, Korkong S, Thongkomplew S, Vichaiwattana P, et al. Hand, Foot and Mouth Disease Caused by Coxsackievirus A6, Thailand, 2012 - Volume 19, Number 4—April 2013 - Emerging Infectious Diseases journal - CDC. . https://wwwnc.cdc.gov/eid/article/19/4/12-1666_article. Accessed 20 May 2020
He Yaqing, Xiao Xinglong. Primer for coxsackie virus A16 nucleic acid detection, probe and kit [Internet]. CN101676406A, 2010. https://patents.google.com/patent/CN101676406A/en. Accessed 21 May 2020
He Yaqing, Xiao Xinglong. Primer for enterovirus 71 type nucleic acid detection, probe and kit [Internet]. CN101676407 A, 2010. http://www.google.com/patents/CN101676407A. Accessed 19 May 2020
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Battistuzzi FU, edtior. Mol Biol Evol 35(6):1547–1549
Yang B, Liu F, Liao Q, Wu P, Chang Z, Huang J, et al. Epidemiology of hand, foot and mouth disease in China, 2008 to 2015 prior to the introduction of EV-A71 vaccine. Euro Surveill [Internet]. 2017. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5743100/. Accessed 22 Apr 2020
Wang J, Hu T, Sun D, Ding S, Carr MJ, Xing W, et al (2017) Epidemiological characteristics of hand, foot, and mouth disease in Shandong, China, 2009–2016. Sci Rep
Wang Y, Feng Z, Yang Y, Self S, Gao Y, Longini IM et al (2011) Hand, foot, and mouth disease in China: patterns of spread and transmissibility. Epidemiology 22(6):781–792
Ji T, Han T, Tan X, Zhu S, Yan D, Yang Q et al (2019) Surveillance, epidemiology, and pathogen spectrum of hand, foot, and mouth disease in mainland of China from 2008 to 2017. Biosafety and Health. 1(1):32–40
Ge S, Yan Q, He S, Zhuang S, Niu J, Xia N (2013) Specific primer amplification of the VP1 region directed by 5′ UTR sequence analysis: Enterovirus testing and identification in clinical samples from hand-foot-and-mouth disease patients. J Virol Methods 193(2):463–469
Cobbin JCA, Britton PN, Burrell R, Thosar D, Selvakumar K, Eden JS et al (2018) A complex mosaic of enteroviruses shapes community-acquired hand, foot and mouth disease transmission and evolution within a single hospital. Virus Evol. 4(2):vey020
Feng X, Guan W, Guo Y, Yu H, Zhang X, Cheng R, et al (2015) A novel recombinant lineage’s contribution to the outbreak of coxsackievirus A6-associated hand, foot and mouth disease in Shanghai, China, 2012-2013. Sci Rep
Xiao K, Duan L, Peng Y, Wu M, Mai G, Yan Z et al (2019) Epidemiologic features of enterovirus associated with hand, foot and mouth disease in 2013 and 2014 in Shenzhen, China. Sci Rep 9(1):3856
Acknowledgements
We acknowledge the Indian Council of Medical Research (ICMR), Government of India, for the grant VIR/NP/66/2013-ECD-1. We are grateful to the District and Taluk hospitals in Kerala and Karnataka for their assistance in clinical and epidemiological data collection.
Author information
Authors and Affiliations
Contributions
Conceptualisation and design of the study by ERS and GA. ERS wrote the first draft of the manuscript and contributed to the data acquisition, analysis, validation, and interpretation. GA supervised the work and acquired the funding. JJ contributed to data analysis. ERS, GA, JJ and SS reviewed and edited the manuscript. SS and CJM did the clinical diagnosis and collection of medical data. SA, SS, KK, CS, VSP, and JA contributed to improve the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Permission to conduct this study was granted by the Institutional Ethics Committee, Kasturba Medical College and Kasturba Hospital (ECR/191/Inst/KL/2013 and ECR/146/Inst/KA/2013/RR-16). All experiments were performed based on the approved guidelines and regulations in the study.
Additional information
Handling Editor: Akbar Dastjerdi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sanjay, R.E., Josmi, J., Sasidharanpillai, S. et al. Molecular epidemiology of enteroviruses associated with hand, foot, and mouth disease in South India from 2015 to 2017. Arch Virol 167, 2229–2238 (2022). https://doi.org/10.1007/s00705-022-05561-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05561-0