
Abstract
Since 2020, SARS-CoV-2 has caused a pandemic virus that has posed many challenges worldwide. Infection with this virus can result in a number of symptoms, one of which is anosmia. Olfactory dysfunction can be a temporary or long-term viral complication caused by a disorder of the olfactory neuroepithelium. Processes such as inflammation, apoptosis, and neuronal damage are involved in the development of SARS-CoV-2-induced anosmia. One of the receptors that play a key role in the entry of SARS-CoV-2 into the host cell is the transmembrane serine protease TMPRSS2, which facilitates this process by cleaving the viral S protein. The gene encoding TMPRSS2 is located on chromosome 21. It contains 15 exons and has many genetic variations, some of which increase the risk of disease. Delta strains have been shown to be more dependent on TMPRSS2 for cell entry than Omicron strains. Blockade of this receptor by serine protease inhibitors such as camostat and nafamostat can be helpful for treating SARS-CoV-2 symptoms, including anosmia. Proper understanding of the different functional aspects of this serine protease can help to overcome the therapeutic challenges of SARS-CoV-2 symptoms, including anosmia. In this review, we describe the cellular and molecular events involved in anosmia induced by SARS-CoV-2 with a focus on the function of the TMPRSS2 receptor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The outbreak of SARS-CoV-2 (COVID-19) originated in Wuhan, China, in December 2019 and spread rapidly across the world, causing a global pandemic [1]. More than 3,000,000 COVID-19 cases were reported worldwide by May 1, 2020. SARS-CoV-2-related olfactory dysfunction has been increasingly recognized as an isolated symptom or in association with other respiratory symptoms [2]. In a recent study, olfactory dysfunction was reported in 45% of COVID-19 patients [3]. Olfactory dysfunction is often transient, with sudden onset in the majority of cases, and average time to recovery is between one and three weeks [4]. There is no significant relationship with sinonasal symptoms, indicating that the pathogenesis of COVID-19-related anosmia might be different from obstructive olfactory dysfunctions that are observed in the setting of other viral upper respiratory tract infections [2, 5]. Although the pathogenesis of COVID-19-related anosmia is not understood, various mechanisms have been proposed. Four different fundamental mechanisms have been proposed regarding smell dysfunction during COVID-19 infection, including infiltration of olfactory centers in the brain, rhinorrhea and nasal congestion and obstruction, damage of olfactory receptor neurons, and injury of supporting cells in the epithelium of the olfactory system. Various viral infections are able to induce nasal congestion, obstruction, and rhinorrhea, which subsequently impede access of odorants to the sensory epithelium and prevent their binding to olfactory receptors [6].
Physical nasal obstruction as a cause of anosmia in COVID-19 [7] has been ruled out due to the absence of nasal obstruction, congestion, and rhinorrhea in a large proportion of the patients with anosmia, who exhibit no mucosal swelling of sinuses or nasal clefts in radiographic imaging [8, 9]. Although sensorineural olfactory loss is considered a possible mechanism for anosmia [10], three major inconsistencies warrant attention, including the absence of viral protein expression, a lack of correlation of cell regeneration with clinical recovery, and a lack of virus in the neurons of the olfactory system. Replacement of olfactory receptor neurons requires 8 to 10 days, plus around five days for maturation of cilia following their death [11], while smell recovery after COVID-19 infection often requires less than seven days [4, 12]. Therefore, functional recovery of anosmia is faster than cilia maturation, neuronal replacement, and growth of neo-axons [6, 13]. Sudden smell loss followed by its rapid recovery is inconsistent with viral infiltration from nose into the olfactory centers of the brain [14, 15]. Currently, there is no evidence available regarding viral access to the brain through the olfactory route during the acute phase of anosmia [2, 16]. Indeed, data derived from genetically modified mouse models are contradictory regarding brain infiltration [17]. The hypothesis of virus-induced damage to support cells in the olfactory epithelium is partly supported by the observation of abundantly expressed entry proteins in sustentacular cells of the olfactory epithelium [18, 19]. However, sustentacular cell death is not necessarily an indication of neural death of olfactory receptors. Rapid smell recovery occurs in parallel with rapid replenishment of sustentacular cells, which is most often observed clinically [6].
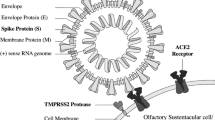
The mechanism by which SARS-CoV-2 enters cells has similarities to those used by influenza virus and human immunodeficiency virus. These viruses have a spike protein (S protein) that belongs to the family of type I viral fusion proteins. The N-terminal subunit of the spike protein (S1) possesses the receptor-binding domain, which binds to the receptor angiotensin-converting enzyme 2 (ACE2) on the host cell, resulting in a conformational change in the S protein, and this is followed by enzymatic cleavage of the S protein by the transmembrane serine protease TMPRSS2 [20, 21]. The C-terminal domain of the spike protein (S2) possesses the heptad repeat domains HR1 and HR2, which facilitate viral entry by forming a six-helix bundle fusion core structure that helps to drive fusion of the viral membrane with the cell membrane [22]. TMPRSS2-induced priming of the S protein and interaction with the ACE2 receptor on the host cell surface are therefore important for viral entry [5, 23].
RNAseq data have implied that the TMPRSS2 gene is expressed in non-neuronal and neuronal cells of the olfactory epithelium [24, 25]. However, in one investigation, no TMPRSS2 expression was observed in the olfactory epithelium [26]. TMPRSS2 expression seems to be higher in non-neuronal olfactory epithelial cells than in olfactory receptor neurons [25, 27]. However, different levels of TMPRSS2 expression are observed in various subpopulations of mature olfactory receptor neurons [25]. Such a mosaic expression pattern in major olfactory receptor neuron genes is not a typical observation. RNAseq profiling has demonstrated that TMPRSS2 is expressed at higher levels in the murine olfactory epithelium than ACE2 [18]. Data from expression profiling of the murine olfactory epithelium indicate that ACE2 is expressed only in non-neuronal cells, whereas TMPRSS2 expression is detected in both non-neuronal and neuronal cells, with possibly higher expression levels in non-neuronal cells [28].
There are numerous candidate drugs with the ability to inhibit SARS-CoV-2 replication and subsequent infection. Among these are inhibitors of ACE2 and inhibitors of TMPRSS2 serine protease. TMPRSS2 blockage might prevent SARS-CoV-2 entry into the cells [29]. Nafamostat mesylate and camostat mesylate are synthetic serine protease inhibitors with the ability to impair viral entry. In a clinical trial involving newly diagnosed outpatients with mild infection, oral camostat mesylate was shown to reduce the viral load of respiratory SARS-CoV-2, leading to rapid resolution of symptoms related to COVID-19 and amelioration of loss of smell and taste [30]. Thus, precise information about the functions of the TMPRSS2 receptor could aid in the development of therapeutic strategies against COVID-19 and its related complications, including anosmia. Here, we describe the molecular and cellular events related to anosmia-induced by SARS-CoV-2, focusing on the function of the TMPRSS2 receptor.
COVID-19 and olfactory disorder
Some of the key symptoms associated with COVID-19, such as fever, chills, cough, difficulty breathing or shortness of breath, sore throat, muscle pain, and sudden loss of taste or smell have also been highlighted previously [31]. Olfactory dysfunction (OD) is characterized by diminished or impaired smell ability while eating (retronasal olfaction) or sniffing (orthonasal olfaction) and is frequently observed in COVID-19 patients with mild symptoms, and even in otherwise asymptomatic carriers. Thus, OD can be considered a possible disease marker, especially in asymptomatic or minimally symptomatic cases [32, 33]. The available data about SARS-CoV-2-related OD indicate a sudden onset of olfactory damage that can occur with or without other signs. Anecdotal reports and unpublished data suggest that olfactory symptoms often resolve after about two weeks, but due to the lack of long-term follow-ups, it is not yet known what percentage of patients experience persistent postinfectious olfactory dysfunction. Coronaviruses are among the pathogens that are known to induce postinfectious OD. Indeed, cells of the nasal epithelium exhibit comparatively high levels of expression of the specific receptors essential for SARS-CoV-2 entry [34, 35]. Disturbance of cell function in the neuroepithelium of olfactory bulbs might lead to inflammatory alterations that impair the functions of olfactory receptor neurons, resulting in impaired neurogenesis and/or improper functioning of these neurons, and these alterations could cause temporary or long-lasting OD [36].
In addition to the functional receptor ACE2, uptake of SARS-CoV-2 is facilitated by transmembrane serine protease 2 (TMPRSS2) [18, 37]. This protein is present in various organs, including heart, lungs, skeletal muscle, oral mucosa, respiratory cells, kidneys, and the central nervous system (CNS). This implies the possibility of multisystem disorders induced by COVID-19 affecting multiple organs simultaneously [14, 38]. Respiratory epithelial cells are the primary site of the attachment of SARS-CoV-2, and it is not particularly surprising that infection with this virus affects the sense of smell and taste [14, 39]. Several possible mechanisms have been proposed for COVID-19-associated anosmia, based on early studies and hospital observations.
The role of inflammation in the development of olfactory disorders induced by SARS-CoV-2
Over time, olfactory neurons begin to die due to ongoing overproduction of cytokines, leading to histological alterations of the neuroepithelium. Notably, when production of inflammatory cytokines is stopped, this is associated with the ability of stem cells in the basal epithelium to regenerate into new olfactory neuronal cells, helping in the recovery of the function of the olfactory system [40]. This suggests that inflammation could result in the development of sensorineural anosmia and that novel therapeutic strategies to eliminate certain inflammatory mediators might be effective in treating SARS-CoV-2-related anosmia [41].
Amongst the cytokines that contribute to inflammatory disorders of the nasal mucosa, tumor necrosis factor (TNF)-α is the most relevant, and it is a powerful mediator of inflammation in various cell types [42, 43]. Although the impact of TNF-α on olfactory neuronal functions is well established, the specific underlying mechanisms of action are not yet well understood. The expression of both TNF-α receptors and mRNA for TNF-α itself in normal epithelial cells has been demonstrated [44], and exposure of olfactory tissue to TNF-α has been associated with specific histopathological alterations. The development of inflammatory infiltration in the setting of local TNF-α expression has been shown to result in a notable expansion of the olfactory submucosa [40].
SARS-CoV-2 infection of the lower or upper respiratory tract causes mild to severe forms of acute respiratory syndrome, which are associated with the release of proinflammatory cytokines such as IL-1β. The binding of SARS-CoV-2 to Toll-like receptors (TLRs) is accompanied by release of pro-IL-1β. Cleavage of pro-IL-1β by caspase-1 leads to activation of inflammasomes and generation of mature IL-1β, which, in turn, is involved in fever, inflammation of lung tissue, and fibrosis. Notably, blocking of members of proinflammatory interleukin-1 family has been shown to be associated with therapeutic benefits with respect to various inflammatory disorders, including viral infections [45]. Torabi et al. [38] demonstrated an increase in the levels of IL-1β in the olfactory epithelium and suggested that little damage to the central nervous system occurred in cases of anosmia because virus was not detected in cerebrospinal fluid in their study. Although different hypotheses have been proposed by different authors regarding the underlying mechanisms involved in anosmia related to COVID-19 [10, 46,47,48,49], the exact mechanism is not known. The data from the above-mentioned study are in line with prior investigations on IL-1β and TNF-α that could be considered when developing treatment and control strategies for SARS-CoV-2 [38].
SARS-CoV-2-infection-associated apoptosis of olfactory cells
Early apoptosis of olfactory cells during viral infection has been studied in animal models. Evidence of apoptosis has been observed in mice with anosmia following intranasal inoculation with some other viruses. Apoptosis and reduced proliferation of olfactory epithelial cells have been demonstrated after intranasal inoculation with Sendai virus (strain 52), which is a mouse counterpart to parainfluenza virus in humans. Another investigation on influenza virus strain R404BP showed apoptosis of neuronal cells in the olfactory system [50]. Inhibited anterograde migration of the virus to the CNS and olfactory bulb has been observed following apoptosis in olfactory cells, and this prevented prolonged olfactory disturbance. This process might be an inherent reaction that prevents a significant course of infection secondary to the regenerative capacity of the neurons of the olfactory system. Viruses with the ability to delay or inhibit apoptosis of olfactory sensory neurons (OSNs) are more prone to enter the brain and olfactory nerve [51, 52].
Levine et al. have commented that neuronal apoptosis is a thorny dilemma, since many important neuronal cells are not renewable during the lifetime of the individual. This limits the advantage of large-scale apoptosis for each kind of respiratory viral infection [53]. This would be a different situation if neurons had self-renewal capacity. Olfactory receptor neurons (ORNs) are a very special exception because of their continual self-renewal every 30-120 days throughout life [54]. Thus, lifelong controlled programmed cell death of ORNs is a usual turnover event. People do not perceive any alteration in the olfactory system when permanent regulated apoptosis is occurring, but intense apoptosis of ORNs can result in sudden loss of the sense of smell. Continuous regeneration of parts of the cranial nerves would make sense teleologically for natural restoration of the sense of smell [55].
It has been demonstrated within last decade that cells of the immune system and cytokines also participate in the regulation of neurogenesis and apoptosis in the neuroepithelium of the olfactory system. It was shown in mice that secretion of growth factors and cytokines and upregulation of ORN regeneration occurred in the presence of activated macrophages, while reduced neurogenesis was seen in their absence [56]. In late 2019, samples obtained from the olfactory mucosa of adult subjects were found to contain numerous immature neuronal cells as well as leukocytes, indicating that neurogenesis in the olfactory system occurs in adults and that immune cells play a fundamental role in homeostasis of the olfactory epithelium [28, 55].
Destruction of olfactory epithelial cells in COVID-19-induced anosmia
Viral infection can lead to partial or total destruction of the nasal olfactory epithelium (OE), including OSNs. In these cases, "postviral anosmia” or “post-upper respiratory infection (URI) anosmia” persists after clearance of rhinitis and associated symptoms of URI for weeks to months until injured regions in the OE of nasal tissue are regenerated. The underlying pathophysiological condition of “postviral anosmia” and related histological examinations have been described in the literature, particularly after infection by rhinoviruses [13, 57]. Neutrophilic inflammation ensues following infection of the nasal respiratory tract and OE, leading to rhinorrhea and mucosal edema. The conductive loss of smell is usually related to the underlying nasal congestion, although in these cases, histological evaluation of the OE revealed the absence of cilia and a loss of some OSNs, which were altered in the metaplastic squamous epithelium, suggesting a sensorineural contribution [5, 58].
Postviral anosmia after human coronavirus 2296 (HCoV-229E) infection has been reported to be associated with olfactory dysfunction lasting more than 6 months [59]. HCoV-229E uses human aminopeptidase N as a receptor for entry into host cells, unlike SARS-CoV-2 and SARS-CoV, which use ACE2 [23, 60]. In SARS-CoV-infected patients, overexpression of ACE2 in the nasal respiratory epithelium has been detected [61], especially on ciliated cells, which is consistent with intranasal entry of the virus [62]. However, these data are in contrast with data from another study in which ACE2 expression was detected in the basal part of the nasal epithelium [63]. Subsequent investigations revealed high expression of ACE2 in goblet cells of the nasal respiratory epithelium [34, 64]. Although goblet cells are absent in the OE [65], recent preliminary studies have found more-prominent ACE2 expression in nonneuronal cells, such as stem cells, supporting cells, and perivascular cells [19]. Specific OE cells co-expressing TMPRSS2 and ACE2 have been identified using a single-cell RNA-seq approach. Preliminary data from a study performed by Fodoulian et al. demonstrated a critical role of sustentacular cells, which face the nasal cavity, in maintenance of the neuroepithelium and as primary cellular targets for entry of SARS-CoV-2 [66]. The high predisposition of nasal tissues to being infected by coronaviruses suggests that some smell loss could be partly attributed to damage to the local environment. COVID-19 patients with slower recovery of olfactory function may have suffered greater intranasal injury. The correlation of long-term olfactory impairment with the severity of clinical signs is not yet clear [5].
Involvement of the nervous system in olfactory impairment caused by COVID-19
Most neurons of the peripheral nervous system (PNS) are involved in the olfactory disorder associated with COVID-19. Signs and symptoms of PNS involvement in SARS-CoV-2 infection, including muscle pain, hypogeusia/ageusia, hyposmia/anosmia, and Guillain-Barre syndrome (GBS), are less severe than those involving the CNS [67]. Ageusia and anosmia are the most common PNS manifestations of SARS-CoV-2 infection, and they have also been observed in infections with other coronaviruses. These sudden-onset symptoms often occur in association with fewer nasal symptoms, including excessive nasal secretion or nasal obstruction [9]. Ageusia and anosmia are often observed in asymptomatic patients or as the initial disease manifestation, with no other symptoms [68]. Thus, it has been suggested by some researchers that individuals with such symptoms could be possible carriers and should be isolated. The sense of taste and smell is gradually regained in most patients after recovery from SARS-CoV-2 infection [69]. An animal study has demonstrated trans-neuronal dissemination of SARS-CoV-2 into the brain through olfactory pathways and its invasion of the olfactory neuroepithelium through the expression of ACE2 and TMPRSS2 in sustentacular cells [70, 71]. Consequently, anosmia occurs due to the disruption of the integrity of the olfactory neuroepithelium. However, anosmia is believed by some authors to be partly due to inflammation in olfactory neurons rather than structural damage to the receptors [72]. Nevertheless, administration of nasal corticosteroids is not yet strongly recommended because of the uncertainty of their benefits [73].
Receptors that affect the sense of smell during SARS-CoV-2 infection
When SARS-CoV-2 enters the olfactory pathway, there is a chance of subsequent brain infection [74]. Thus, it might be expected that olfactory epithelial cells express proteins that facilitate the entry of SARS-CoV-2. Presently, this is unknown, but if certain tissues are found to have a high viral load, this might be useful for diagnosis of infection in individuals with no symptoms [75]. Invasion of human cells by SARS-CoV-2 virus via ACE2 as its obligatory receptor facilitates further viral uptake by TMPRSS2, a priming protease [23]. Cells with high expression level of TMPRSS2 and ACE2 demonstrate strong viral attachment affinity and are more prone to infection [76], and identification of the relevant cell types is important [75].
Although transcriptomic investigations have evaluated gene expression profiles in the epithelium of the olfactory system in different species, expression of TMPRSS2 and ACE2 in neurons and nonneuronal cells present in the epithelium and their age-dependent expression level remain controversial [25, 27, 77]. In an in vivo animal model, high expression levels of TMPRSS2 and ACE2 were observed in sustentacular cells using in situ hybridization, RT-PCR, RNAseq, immunocytochemistry, and western blot, suggesting these cells as possible targets of SARS-CoV-2 in the epithelium of the human olfactory system [18]. Since sustentacular cells play a fundamental role in supportive metabolism of olfactory neurons and the sense of smell [78], COVID-19-induced damage to sustentacular cells has been proposed to result in olfactory damage in COVID-19 patients [18].
The TMPRSS2 and its role in the olfactory system
TMPRSS2 gene is conserved in frogs, rhesus monkeys, chimpanzees, cows, dogs, rats, mice, chickens, zebrafish, and C. elegans. In humans, it is located at 21q22.3 and possesses an open reading frame with 15 exons encoding 492 residues. It has a scavenger receptor cysteine-rich (SRCR) domain, which contributes to its attachment to extracellular molecules or other cell surfaces, an LDL receptor class A (LDLRA) domain, which is a calcium-binding site, a type II transmembrane domain, and a serine protease domain of the S1 family, which cuts at lysine or arginine residues. Furthermore, TMPRSS2 contains an androgen response element upstream of the coding region. Dihydrotestosterone and testosterone are potent transcriptional regulators of this gene via the androgen receptor [79].
A disulfide bond links the membrane-bound portion of this protein with the catalytic domain, which is positioned in the extracellular region. The N-terminal domain is located in the intracellular region, and it is followed by a stem region, a transmembrane region, and a protease domain containing the catalytic triad of serine, histidine, and aspartate necessary for cleavage activity [80]. Interaction of the intracellular domain with components of the cytoskeleton and signaling molecules is important for intracellular peptide trafficking. The stem region, which contains the SRCR and LDLRA domains, facilitates protein-protein interactions. The enzymatic region is able to cleave receptors of the cell membrane, growth factors, cytokines, and extracellular matrix components. Following autocleavage, the serine protease domain is secreted into the epithelium and subsequently interacts with extracellular matrix proteins on the cell surface and proteins of neighboring cells [81].
The isoform of TMPRSS2 containing 492 amino acids is called isoform 2. Alternative mRNA splicing results in formation of isoform 1, which is similar to isoform 2 with the difference that it has an extra N-terminal cytoplasmic domain and has been observed to be expressed in lung-related tissues. Both of these molecules are activated autocatalytically, indicating that cleavage of the proenzyme occurs between the carboxyl-end catalytic region and the rest of the molecule. This leads to conformational changes in the protease domain that are essential for conversion to its active form. The generation of alternative isoforms results in circulating and membrane-bound forms. The presence of a single N-terminal fragment and two fragments are characteristic of isoforms 2 and 1, respectively, suggesting potential differences in cleavage specificity and, possibly, intracellular localization [82]. The major fraction of the mature protease after autocatalytic cleavage is membrane-bound; however, a portion is also present in the extracellular matrix. Isoform 1 is colocalized with the viral haemagglutinin at the site where it is activated by proteolytic cleavage. Isoform 1 also activates the SARS-CoV S protein, allowing the virus to enter target cells through a cathepsin-L-independent pathway [83].
Among members of the type II TTSP family, TMPRSS2 is unusual because of its participation in various complexes. Most of the TMPRSS2 receptor is released from zymogen complexes, which do not appear to be stable despite the presence of endogenous protease inhibitors. Therefore, complexes containing TMPRSS2 do not seem to be by-products of specific mechanisms for prevention of zymogen activation. TMPRSS2 contains 22 cysteine residues, and its serine protease domain includes eight cysteines in its conserved regions. Cys140 in the serine protease domain is unpaired and therefore has the potential to participate in the formation of disulfide-linked complexes. This is a potentially important observation suggesting a novel mechanism by which protease activity can be regulated by complex formation [80, 84, 85].
Binding of ACE2 to the receptor‐binding domain (RBD) of the S protein initiates viral infection. It has been hypothesized that the high rate of infection and transmissibility of SARS‐CoV‐2 compared to other coronaviruses such as SARS‐CoV might be attributed to high affinity of the S protein for the ACE2 receptor [86,87,88]. However, the binding of the RBD domain of SARS-CoV-2 to ACE2 is less efficient than that of SARS‐CoV [89], suggesting the involvement of other factors. Thus, the virus might have undergone natural selection to overcome its low affinity for ACE2 [90], as coronaviruses are able to specifically adapt to a host due to their high genomic plasticity [91]. The S protein of SARS‐CoV‐2 is cleaved by TMPRSS2 after it binds to the ACE2 receptor. Cleavage at the S1/S2 and S2 sites is important for fusion of the viral membrane with the membrane of the host cell [23, 92]. In the absence of TMPRSS2 receptors, SARS‐CoV‐2 appears to use other proteases, including cathepsin B/L, which is present in lung tissue but is not able to substitute for TMPRSS2 in the case of MERS‐CoV and SARS‐CoV [93].
Anosmia is speculated to be associated with COVID-19-induced damage to the epithelium and its consequent inflammation and/or malfunction of neuronal receptors located in the olfactory structure. The latter is of great importance, since SARS‐CoV has been shown to infect brain tissue of transgenic mice expressing human ACE2 protein following its initial impact on olfactory receptors [94]. If this also applies to ASRS-CoV-2, the olfactory epithelium requires TMPRSS2- and ACE2-expressing cells, facilitating viral infection [75, 76]. To investigate this, Bilinska et al. examined TMPRSS2 and ACE2 expression in mouse olfactory epithelial cells [18]. They found that expression of both enzymes could be observed in sustentacular OE cells. While ACE2 expression was not detected in olfactory receptor neurons (ORNs), a low level of TMPRSS2 was expressed in mature ORNs. The preferred targeting of sustentacular cells by SARS-CoV-2 results in buildup of infected cells, which interferes with their metabolism. The improper functioning of sustentacular cells could partly explain the loss of olfaction due to the importance of these cells for olfaction by endocytosis of olfactory binding protein‐odorant complexes and secretion of odor-binding proteins [95]. However, this could not explain the ability of SARS‐CoV‐2 to target the cells of brain tissue. Additional investigation is required to determine if SARS-CoV-2 is able to infect ORNs on the way to subsequently infecting the brain [18]. Murine OE cells have been reported by Bilinska et al. [18] to contain larger amounts of ACE2 proteins than respiratory epithelial cells, making OE cells more prone to being infected by SARS‐CoV‐2 than the cells in the respiratory epithelium. This effect can be simulated in human OE and respiratory epithelial cells to compare levels of TMPRSS2 and ACE2. If this observation also applies to human OE cells, it might be more appropriate to test for SARS‐CoV‐2 in the OE than in the respiratory epithelium, potentially reducing the likelihood of obtaining a false‐negative test result for COVID‐19 [96].
Interaction of different strains of SARS-CoV-2 with the TMPRSS2 receptor
The increased transmission efficiency of the Omicron variant of SARS-CoV-2 has caused great concern. Zhao et al. showed that the replication and fusion activity of the Delta variant is significantly increased in VeroE6/TMPRSS2 cells, while the fusion and replication of the Omicron variant are much less dependent on TMPRSS2 [97]. For fusion of the cellular and viral membranes, a conformational change in the spike protein is required [98]. After cleavage at the S1/S2 junction by furin, the S2’ site can be cleaved by either endosomal cathepsin B/L or cell-surface TMPRSS2. High TMPRSS2 expression was demonstrated in lung alveolar cells through single-cell sequencing [34]. TMPRSS2-enhanced replication of the Delta strain is correlated with earlier in vivo investigations indicating more-extensive involvement of alveolar pneumocytes in infection by the Delta variant [99]. Zhao et al. demonstrated a higher dependence of the Omicron variant on TMPRSS2 in comparison to the Delta variant and suggested possible poorer replication of the Omicron variant in the lungs than the Delta variant [97].
Preliminary epidemiological studies have suggested that the Omicron variant causes milder disease [97]. The Delta and Omicron variants both use the same S2’ cleavage site, but there is nevertheless a marked difference in TMPRSS2 dependence for viral replication, which might be attributed to a difference in the cleavage site for furin, which is P681H for Omicron and P681R for Delta. Using a pseudovirus system, Peacock et al. demonstrated a much higher efficiency of TMPRSS2-mediated entry of pseudoviruses carrying a polybasic furin cleavage site at the S1/S2 junction than of pseudoviruses with a deletion of this cleavage site [100]. The Omicron variant has been shown by Zhao et al. to be much less fusogenic than the Delta variant. This feature could be due to the difference in the furin cleavage site at the S1/S2 junction as well as the difference in TMPRSS2 dependence. The Delta variant was shown previously to be more fusogenic than the wild-type or Alpha variant containing the P681H mutation [97, 99, 101, 102]. The observation of syncytia in postmortem lung samples of expired SARS-CoV-2 patients has suggested an association of the fusion acuity of virus with the severity of disease [103]. In addition to cleaving the spike protein for its activation, TMPRSS2 plays role as an interferon antagonist, and TMPRSS2 overexpression is able to reverse restriction of replication of SARS-CoV-2 by NCOA7, which is an interferon-stimulated gene [104].
Further investigations evaluating how viral replication is affected by TMPRSS2 in the context of the innate immune response will be of great interest. Although a higher level of expression of TMRPSS2 has been observed in both Calu3 and VeroE6/TMPRSS2 cells, the Omicron variant exhibits different replication efficiency in these cell lines. While Omicron can replicate to the same level as the Delta variant in VeroE6/TMPRSS2 cells at 48 and 72 hpi, it reaches a significantly lower level than the Delta variant in Calu3 cells at 48 and 72 hpi. This difference could be due to the fact that SARS-CoV-2 does not use endocytosis to enter Calu3 cells [97]. A study has demonstrated the inability of chloroquine, which is an inhibitor of endosomal acidification, to prevent replication of SARS-CoV-2 in Calu3 cells [105], whereas another study showed inhibition by chloroquine in VeroE6/TMPRSS2 cells, indicating viral entry through the endosomal pathway [97]. Further studies using animal models are needed to determine whether differences in TMPRSS2 dependence result in different tissue tropism or disease severity [97].
Risk of COVID-19 and TMPRSS2 gene polymorphisms
There is a non-uniform impact of the COVID-19 crisis across ethnic groups, with certain groups affected disproportionately [106]. Discrepancies in the rate of infection and case fatality rate could be attributed to multiple causes, including underlying comorbidities, differences in access to medical care, social distancing policies, population age structure and coverage, and reliability of epidemiological data demonstrating higher mortality in the elderly population and individuals with underlying comorbidities [107, 108]. However, there have been many deaths of young and healthy individuals due to rapid cytokine storms [109]. Although this is not the whole story, and not all of the disparities among groups can be explained by these factors, data from countries that apply strict standards for gathering and presenting epidemiological data suggest that variations in human genetic makeup might explain differences in susceptibility to and severity of disease in different populations [106], and there is evidence supporting a role of variations in the TMPRSS2 and ACE2 genes in susceptibility to COVID-19 among different populations [110, 111].
Within the human TMPRSS2 gene, numerous single-nucleotide polymorphisms (SNPs) have been identified in a recent analysis using computational modeling (dbSNP, NCBI). Of these, just 21 variations with minor allele frequency of 0.01-0.95 were found to affect enzyme function [112], and only two of those are missense variations (rs75603675 and rs12329760). In several investigations, the rs12329760 polymorphism, which is referred to as the p.Val160Met variant, has been demonstrated to be associated with the risk of prostate cancer. This could confirm the clinical consequences related to this genetic variant [113,114,115,116,117]. This genetic variation is located on exon 6 and could alter the function of the gene (Figs. 1 and 2). Although it affects gene expression, it has no effect on the mRNA structure or function of TMPRSS2 (Fig. 2). Various effects of TMPRSS2 gene polymorphisms have been presented in our previous publications [118,119,120,121,122,123,124,125,126], but additional SNPs have been found to have an impact on the risk of COVID-19. These TMPRSS2 gene polymorphisms and their association with the risk of COVID-19 are listed in Table 1.

TMPRSS2 gene map and features of two common gene variations, rs12329760 and rs75603675. The TMPRSS2 gene contains 15 exons, and rs12329760 and rs75603675 are located on exons 1 and 6, respectively. The position of the rs12329760 polymorphism in the three-dimensional structure of the protein suggests an effect on enzyme function.

Effects of the rs12329760 polymorphism on protein and mRNA function and gene expression. A Potential deleterious impact of rs12329760 on the TMPRSS2 protein. B RNAsnp analysis showing no impact on mRNA structure. C Effect of this genetic variation on TMPRSS2 expression.
Therapeutic strategies targeting the TMPRSS2 receptor
Multiple mechanisms involved in the replication and infectivity of SARS-CoV-2 provide potential targets for pharmacological interventions. Infection of macrophages, pneumocytes, and pulmonary mast cells requires the viral S protein. The entry pathway involving binding of the S protein to the ACE2 cell receptor is mediated through host-cell-derived TMPRSS2 serine protease [23]. Viral RNA release, replication, and translation occur after viral entry into the host cell. Translated viral polyproteins are eventually cleaved by viral proteases to form mature effector proteins [133]. Viral infection is initiated by the interaction of viral S protein with ACE2 on cytoplasmic membrane of the host cell. Numerous candidate drugs have the potential to inhibit SARS-CoV-2 replication and subsequent rounds of infection. Such drugs included ACE2 inhibitors and inhibitors of TMPRSS2 serine protease. Blocking of TMPRSS2, which is essential for priming of the S protein, and inhibition of ACE2, which is the host cell receptor of SARS-CoV-2, might prevent cellular entry of SARS-CoV-2 [29].
Shen et al. demonstrated the importance of a guanine-rich tract in the human TMPRSS2 promoter for formation of an intramolecular G-quadruplex structure in the presence of K+ for gene transcriptional activity. In order to stabilize G-quadruplexes, seven novel benzoselenoxanthene analogues have been introduced. Compounds that downregulate TMPRSS2 gene expression have been shown to suppress propagation of influenza A virus in vitro [134]. Thus, small molecules that target the TMPRSS2 gene G-quadruplex and subsequently inhibit TMPRSS2 expression provide a novel strategy against influenza A virus. This could also be a potential anti-SARS-CoV-2 target [134].
Numerous viruses employ host cell proteases to activate their envelope glycoproteins, including MERS coronavirus (MERS-CoV), Ebola virus, influenza virus, and SARS coronavirus (SARS-CoV) [61, 135, 136]. Cleavage of the viral spike protein and its subsequent activation are required for membrane fusion and entry into the host cell, and this is facilitated by TMPRSS2 [21, 23, 137, 138]. Figure 3 shows how the administration of two serine protease inhibitors, camostat and nafamostat, can prevent the virus from entering the cell. Camostat mesylate, a commercial serine protease inhibitor is able to partially block HCoV-NL63 and SARS-CoV entry in TMPRSS2- and ACE2-expressing HeLa cells [139]. Indeed, inhibition of TMPRSS2 in human lung Calu-3 cells using camostat mesylate was shown to be associated with significantly diminished SARS-CoV-2 infection [23].

Inhibitory effect of camostat and nafamostat on SARS-CoV-2 entry into the cell. Because TMPRSS2 plays a key role in virus entry, the serine protease inhibitors camostat and nafamostat can inhibit virus cell entry by blocking TMPRSS2.
Camostat and camostat mesylate are synthetic serine protease inhibitors that were developed decades ago to treat dystrophic epidermolysis [140], oral squamous cell carcinoma [141, 142], and chronic pancreatitis [143,144,145], to inhibit and exocrine pancreatic enzymes [146, 147]. Camostat mesylate (NI-03), prescribed three times per day at the recommended dose of 100–300 mg, has been manufactured by Nichi-Iko Pharmaceutical Co., Ltd. and Ono Pharmaceutical, Japan, as an oral drug [145]. A clinical trial on 95 patients who received 200 mg of camostat mesylate three times per day for 2 weeks for the treatment of dyspepsia associated with non-alcoholic mild pancreatic disease demonstrated only mild adverse effects [143], indicating that this drug is well tolerated [29]. A clinical trial demonstrated a more rapid resolution of COVID-19 symptoms and amelioration of lost taste and smell in outpatients with newly diagnosed mild COVID-19 infection who received oral camostat mesylate [30]. Nafamostat mesylate is a synthetic serine protease that is clinically approved in Japan to treat disseminated intravascular coagulation and acute pancreatitis. It is also prescribed as an anticoagulant for extracorporeal circulation [148,149,150]. In a study screening about 1100 FDA-approved drugs, nafamostat mesylate was found to prevent MERS-CoV S-protein-mediated viral membrane fusion with TMPRSS2-expressing lung Calu-3 host cells by inhibiting TMPRSS2 protease activity [151]. Since the S proteins of SARS-CoV-2 and MERS-Cov share considerable amino acid sequence similarity [152, 153], nafamostat mesylate could potentially inhibit cellular entry of SARS-CoV-2. In a cell culture study on simian Vero E6 cells infected with SARS-CoV-2, nafamostat mesylate was found to have inhibitor activity, with an EC50 of 22.50 μM [154], suggesting its ability to prevent SARS-CoV-2 infection. In a phase II, randomized, multicenter, open-label trial including 19 subjects with a severe form of acute pancreatitis, daily intravenous administration of nafamostat mesylate at a dose of 240 mg for five consecutive days showed benefits without significant adverse effects [150].
Conclusion
A significantly high proportion of patients with COVID-19 have symptoms of anosmia, but the underlying molecular and cellular mechanisms remain unclear. Upper respiratory infection manifests classically as nasal obstruction and rhinorrhea, leading to conductive olfactory loss. Postviral anosmia might ensue in a subacute form after resolution of acute symptoms of upper respiratory tract infection. However, preliminary data regarding COVID-19 cases have revealed a novel syndrome of acute-onset anosmia without nasal obstruction or rhinitis. New studies have revealed the identity of the cells responsible for viral entry into the olfactory neural system. Reviewing the literature on anosmia induced by viral infection allows specific mechanisms of anosmia to be postulated, but the exact mechanisms are not yet clear. Several hypotheses have been proposed for COVID-19-associated anosmia. According to an animal study, coronaviruses are able to disseminate transneuronally into the brain using olfactory pathways and invade the olfactory neuroepithelium in a manner dependent on the expression of ACE2 and TMPRSS2 in sustentacular cells. Because of its important role in viral entry, information about the function of TMPRSS2 could suggest therapeutic strategies against COVID-19 and its complications, including anosmia.
References
Veronese S, Sbarbati A (2021) Chemosensory systems in COVID-19: evolution of scientific research. ACS Chem Neurosci 12(5):813–824. https://doi.org/10.1021/acschemneuro.0c00788
Cooper KW, Brann DH, Farruggia MC, Bhutani S, Pellegrino R, Tsukahara T et al (2020) COVID-19 and the chemical senses: supporting players take center stage. Neuron 107(2):219–233. https://doi.org/10.1016/j.neuron.2020.06.032
Hoang MP, Kanjanaumporn J, Aeumjaturapat S, Chusakul S, Seresirikachorn K, Snidvongs K (2020) Olfactory and gustatory dysfunctions in COVID-19 patients: a systematic review and meta-analysis. Asian Pac J Allergy Immunol 38(3):162–169. https://doi.org/10.12932/ap-210520-0853
Lee Y, Min P, Lee S, Kim SW (2020) Prevalence and duration of acute loss of smell or taste in COVID-19 patients. J Korean Med Sci 35(18):e174. https://doi.org/10.3346/jkms.2020.35.e174
Han AY, Mukdad L, Long JL, Lopez IA (2020) Anosmia in COVID-19: mechanisms and significance. Chem Senses. https://doi.org/10.1093/chemse/bjaa040
Butowt R, von Bartheld CS (2021) Anosmia in COVID-19: underlying mechanisms and assessment of an olfactory route to brain infection. Neuroscientist 27(6):582–603. https://doi.org/10.1177/1073858420956905
Eliezer M, Hautefort C, Hamel AL, Verillaud B, Herman P, Houdart E et al (2020) Sudden and complete olfactory loss of function as a possible symptom of COVID-19. JAMA Otolaryngol Head Neck Surg 146(7):674–675. https://doi.org/10.1001/jamaoto.2020.0832
Printza A, Constantinidis J (2020) The role of self-reported smell and taste disorders in suspected COVID-19. Eur Arch Oto-rhino-laryngol 277(9):2625–2630. https://doi.org/10.1007/s00405-020-06069-6
Vaira LA, Salzano G, Deiana G, De Riu G (2020) Anosmia and ageusia: common findings in COVID-19 patients. Laryngoscope 130(7):1787. https://doi.org/10.1002/lary.28692
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11(7):995–998. https://doi.org/10.1021/acschemneuro.0c00122
Brann JH, Firestein SJ (2014) A lifetime of neurogenesis in the olfactory system. Front Neurosci 8:182. https://doi.org/10.3389/fnins.2014.00182
Sedaghat AR, Gengler I, Speth MM (2020) Olfactory dysfunction: a highly prevalent symptom of COVID-19 with public health significance. Otolaryngology Head Neck Surg 163(1):12–15. https://doi.org/10.1177/0194599820926464
Bryche B, St Albin A, Murri S, Lacôte S, Pulido C, Ar Gouilh M et al (2020) Massive transient damage of the olfactory epithelium associated with infection of sustentacular cells by SARS-CoV-2 in golden Syrian hamsters. Brain Behav Immun 89:579–586. https://doi.org/10.1016/j.bbi.2020.06.032
Aragão M, Leal MC, Cartaxo Filho OQ, Fonseca TM, Valença MM (2020) Anosmia in COVID-19 associated with injury to the olfactory bulbs evident on MRI. AJNR Am J Neuroradiol 41(9):1703–1706. https://doi.org/10.3174/ajnr.A6675
Politi LS, Salsano E, Grimaldi M (2020) Magnetic resonance imaging alteration of the brain in a patient with Coronavirus disease 2019 (COVID-19) and anosmia. JAMA Neurol 77(8):1028–1029. https://doi.org/10.1001/jamaneurol.2020.2125
Wang L, Shen Y, Li M, Chuang H, Ye Y, Zhao H et al (2020) Clinical manifestations and evidence of neurological involvement in 2019 novel coronavirus SARS-CoV-2: a systematic review and meta-analysis. J Neurol 267(10):2777–2789. https://doi.org/10.1007/s00415-020-09974-2
Bao L, Deng W, Huang B, Gao H, Liu J, Ren L et al (2020) The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583(7818):830–833. https://doi.org/10.1038/s41586-020-2312-y
Bilinska K, Jakubowska P, Von Bartheld CS, Butowt R (2020) Expression of the SARS-CoV-2 Entry Proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: identification of cell types and trends with age. ACS Chem Neurosci 11(11):1555–1562. https://doi.org/10.1021/acschemneuro.0c00210
Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B et al (2020) Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv. https://doi.org/10.1126/sciadv.abc5801
Chen J, Subbarao K (2007) The Immunobiology of SARS*. Annu Rev Immunol 25:443–472. https://doi.org/10.1146/annurev.immunol.25.022106.141706
Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T (2011) A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol 85(2):873–882. https://doi.org/10.1128/jvi.02062-10
Du L, He Y, Zhou Y, Liu S, Zheng BJ, Jiang S (2009) The spike protein of SARS-CoV–a target for vaccine and therapeutic development. Nat Rev Microbiol 7(3):226–236. https://doi.org/10.1038/nrmicro2090
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–80.e8. https://doi.org/10.1016/j.cell.2020.02.052
Kanageswaran N, Demond M, Nagel M, Schreiner BS, Baumgart S, Scholz P et al (2015) Deep sequencing of the murine olfactory receptor neuron transcriptome. PLoS ONE 10(1):e0113170. https://doi.org/10.1371/journal.pone.0113170
Saraiva LR, Ibarra-Soria X, Khan M, Omura M, Scialdone A, Mombaerts P et al (2015) Hierarchical deconstruction of mouse olfactory sensory neurons: from whole mucosa to single-cell RNA-seq. Sci Rep 5:18178. https://doi.org/10.1038/srep18178
Olender T, Keydar I, Pinto JM, Tatarskyy P, Alkelai A, Chien MS et al (2016) The human olfactory transcriptome. BMC Genom 17(1):619. https://doi.org/10.1186/s12864-016-2960-3
Nickell MD, Breheny P, Stromberg AJ, McClintock TS (2012) Genomics of mature and immature olfactory sensory neurons. J Comp Neurol 520(12):2608–2629. https://doi.org/10.1002/cne.23052
Durante MA, Kurtenbach S, Sargi ZB, Harbour JW, Choi R, Kurtenbach S et al (2020) Single-cell analysis of olfactory neurogenesis and differentiation in adult humans. Nat Neurosci 23(3):323–326. https://doi.org/10.1038/s41593-020-0587-9
McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C (2020) Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res 157:104859. https://doi.org/10.1016/j.phrs.2020.104859
Chupp G, Spichler-Moffarah A, Søgaard OS, Esserman D, Dziura J, Danzig L et al (2022) A phase 2 randomized, double-blind, placebo-controlled trial of oral camostat mesylate for early treatment of COVID-19 outpatients showed shorter illness course and attenuation of loss of smell and taste. medRxiv. https://doi.org/10.1101/2022.01.28.22270035
Koyama S, Ueha R, Kondo K (2021) Loss of smell and taste in patients with suspected COVID-19: analyses of patients’ reports on social media. J Med Internet Res 23(4):e26459. https://doi.org/10.2196/26459
Spinato G, Fabbris C, Polesel J, Cazzador D, Borsetto D, Hopkins C et al (2020) Alterations in smell or taste in mildly symptomatic outpatients with SARS-CoV-2 infection. JAMA 323(20):2089–2090. https://doi.org/10.1001/jama.2020.6771
Desai M, Oppenheimer J (2021) The importance of considering olfactory dysfunction during the COVID-19 pandemic and in clinical practice. J Allergy Clin Immunol Pract 9(1):7–12. https://doi.org/10.1016/j.jaip.2020.10.036
Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M et al (2020) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med 26(5):681–687. https://doi.org/10.1038/s41591-020-0868-6
Rebholz H, Pfaffeneder-Mantai F, Knoll W, Hassel AW, Frank W, Kleber C (2021) Olfactory dysfunction in SARS-CoV-2 infection: Focus on odorant specificity and chronic persistence. Am J Otolaryngol 42(5):103014. https://doi.org/10.1016/j.amjoto.2021.103014
Whitcroft KL, Hummel T (2020) Olfactory dysfunction in COVID-19: diagnosis and management. JAMA 323(24):2512–2514. https://doi.org/10.1001/jama.2020.8391
Sayin I, Yazici ZM (2020) Taste and smell impairment in SARS-CoV-2 recovers early and spontaneously: experimental data strongly linked to clinical data. ACS Chem Neurosci 11(14):2031–2033. https://doi.org/10.1021/acschemneuro.0c00296
Torabi A, Mohammadbagheri E, Akbari Dilmaghani N, Bayat AH, Fathi M, Vakili K et al (2020) Proinflammatory cytokines in the olfactory mucosa result in COVID-19 induced anosmia. ACS Chem Neurosci 11(13):1909–1913. https://doi.org/10.1021/acschemneuro.0c00249
Dushianthan A, Clark H, Madsen J, Mogg R, Matthews L, Berry L et al (2020) Nebulised surfactant for the treatment of severe COVID-19 in adults (COV-Surf): a structured summary of a study protocol for a randomized controlled trial. Trials 21(1):1014. https://doi.org/10.1186/s13063-020-04944-5
Lane AP, Turner J, May L, Reed R (2010) A genetic model of chronic rhinosinusitis-associated olfactory inflammation reveals reversible functional impairment and dramatic neuroepithelial reorganization. J Neurosci 30(6):2324–2329. https://doi.org/10.1523/jneurosci.4507-09.2010
Goncalves S, Goldstein BJ (2016) Pathophysiology of olfactory disorders and potential treatment strategies. Curr Otorhinolaryngol Rep 4(2):115–121. https://doi.org/10.1007/s40136-016-0113-5
Kollias G, Douni E, Kassiotis G, Kontoyiannis D (1999) On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol Rev 169:175–194. https://doi.org/10.1111/j.1600-065x.1999.tb01315.x
Sandborn WJ, Hanauer SB (1999) Antitumor necrosis factor therapy for inflammatory bowel disease: a review of agents, pharmacology, clinical results, and safety. Inflamm Bowel Dis 5(2):119–133. https://doi.org/10.1097/00054725-199905000-00008
Suzuki Y, Farbman AI (2000) Tumor necrosis factor-alpha-induced apoptosis in olfactory epithelium in vitro: possible roles of caspase 1 (ICE), caspase 2 (ICH-1), and caspase 3 (CPP32). Exp Neurol 165(1):35–45. https://doi.org/10.1006/exnr.2000.7465
Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I et al (2020) Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents 34(2):327–331. https://doi.org/10.23812/conti-e
Soler ZM, Patel ZM, Turner JH, Holbrook EH (2020) A primer on viral-associated olfactory loss in the era of COVID-19. Int Forum Allergy Rhinol 10(7):814–820. https://doi.org/10.1002/alr.22578
Vaira LA, Salzano G, De Riu G (2020) The importance of olfactory and gustatory disorders as early symptoms of coronavirus disease (COVID-19). Br J Oral Maxillofac Surg 58(5):615–616. https://doi.org/10.1016/j.bjoms.2020.04.024
Lechien JR, Hopkins C, Saussez S (2020) Sniffing out the evidence; It’s now time for public health bodies recognize the link between COVID-19 and smell and taste disturbance. Rhinology 58(4):402–403. https://doi.org/10.4193/Rhin20.159
Gilani S, Roditi R, Naraghi M (2020) COVID-19 and anosmia in Tehran, Iran. Med Hypotheses 141:109757. https://doi.org/10.1016/j.mehy.2020.109757
Imam SA, Lao WP, Reddy P, Nguyen SA, Schlosser RJ (2020) Is SARS-CoV-2 (COVID-19) postviral olfactory dysfunction (PVOD) different from other PVOD? World J Otorhinolaryngol - Head Neck Surg 6(Suppl 1):S26-s32. https://doi.org/10.1016/j.wjorl.2020.05.004
van Riel D, Verdijk R, Kuiken T (2015) The olfactory nerve: a shortcut for influenza and other viral diseases into the central nervous system. J Pathol 235(2):277–287. https://doi.org/10.1002/path.4461
Najafloo R, Majidi J, Asghari A, Aleemardani M, Kamrava SK, Simorgh S et al (2021) Mechanism of anosmia caused by symptoms of COVID-19 and emerging treatments. ACS Chem Neurosci 12(20):3795–3805. https://doi.org/10.1021/acschemneuro.1c00477
Levine B (2002) Apoptosis in viral infections of neurons: a protective or pathologic host response? Curr Top Microbiol Immunol 265:95–118. https://doi.org/10.1007/978-3-662-09525-6_5
Mori I (2015) Transolfactory neuroinvasion by viruses threatens the human brain. Acta Virol 59(4):338–349. https://doi.org/10.4149/av_2015_04_338
Le Bon SD, Horoi M (2020) Is anosmia the price to pay in an immune-induced scorched-earth policy against COVID-19? Med Hypotheses 143:109881. https://doi.org/10.1016/j.mehy.2020.109881
Borders AS, Getchell ML, Etscheidt JT, van Rooijen N, Cohen DA, Getchell TV (2007) Macrophage depletion in the murine olfactory epithelium leads to increased neuronal death and decreased neurogenesis. J Comp Neurol 501(2):206–218. https://doi.org/10.1002/cne.21252
Othman BA, Maulud SQ, Jalal PJ, Abdulkareem SM, Ahmed JQ, Dhawan M et al (2012) Olfactory dysfunction as a post-infectious symptom of SARS-CoV-2 infection. Ann Med Surg 2022(75):103352. https://doi.org/10.1016/j.amsu.2022.103352
Yamagishi M, Fujiwara M, Nakamura H (1994) Olfactory mucosal findings and clinical course in patients with olfactory disorders following upper respiratory viral infection. Rhinology 32(3):113–118
Suzuki M, Saito K, Min WP, Vladau C, Toida K, Itoh H et al (2007) Identification of viruses in patients with postviral olfactory dysfunction. Laryngoscope 117(2):272–277. https://doi.org/10.1097/01.mlg.0000249922.37381.1e
Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT et al (1992) Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357(6377):420–422. https://doi.org/10.1038/357420a0
Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P et al (2012) Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 7(4):e35876. https://doi.org/10.1371/journal.pone.0035876
Sims AC, Baric RS, Yount B, Burkett SE, Collins PL, Pickles RJ (2005) Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J Virol 79(24):15511–15524. https://doi.org/10.1128/jvi.79.24.15511-15524.2005
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203(2):631–637. https://doi.org/10.1002/path.1570
Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN et al (2020) SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181(5):1016–35.e19. https://doi.org/10.1016/j.cell.2020.04.035
Solbu TT, Holen T (2012) Aquaporin pathways and mucin secretion of Bowman’s glands might protect the olfactory mucosa. Chem Senses 37(1):35–46. https://doi.org/10.1093/chemse/bjr063
Fodoulian L, Tuberosa J, Rossier D, Boillat M, Kan C, Pauli V et al (2020) SARS-CoV-2 receptors and entry genes are expressed in the human olfactory neuroepithelium and brain. iScience 23(12):101839. https://doi.org/10.1016/j.isci.2020.101839
Sedaghat Z, Karimi N (2020) Guillain Barre syndrome associated with COVID-19 infection: a case report. J Clin Neurosci 76:233–235. https://doi.org/10.1016/j.jocn.2020.04.062
Gane SB, Kelly C, Hopkins C (2020) Isolated sudden onset anosmia in COVID-19 infection. A novel syndrome? Rhinology 58(3):299–301. https://doi.org/10.4193/Rhin20.114
Niazkar HR, Zibaee B, Nasimi A, Bahri N (2020) The neurological manifestations of COVID-19: a review article. Neurol Sci 41(7):1667–1671. https://doi.org/10.1007/s10072-020-04486-3
Xydakis MS, Dehgani-Mobaraki P, Holbrook EH, Geisthoff UW, Bauer C, Hautefort C et al (2020) Smell and taste dysfunction in patients with COVID-19. Lancet Infect Dis 20(9):1015–1016. https://doi.org/10.1016/s1473-3099(20)30293-0
Moein ST, Hashemian SM, Mansourafshar B, Khorram-Tousi A, Tabarsi P, Doty RL (2020) Smell dysfunction: a biomarker for COVID-19. Int Forum Allergy Rhinol 10(8):944–950. https://doi.org/10.1002/alr.22587
Lorenzo Villalba N, Maouche Y, Alonso Ortiz MB, Cordoba Sosa Z, Chahbazian JB, Syrovatkova A et al (2020) Anosmia and dysgeusia in the absence of other respiratory diseases: should COVID-19 infection be considered? Eur J Case Rep Intern Med 7(4):001641. https://doi.org/10.12890/2020_001641
Rashid RA, Zgair A, Al-Ani RM (2021) Effect of nasal corticosteroid in the treatment of anosmia due to COVID-19: a randomised double-blind placebo-controlled study. Am J Otolaryngol 42(5):103033. https://doi.org/10.1016/j.amjoto.2021.103033
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q et al (2020) Neurologic manifestations of hospitalized patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol 77(6):683–690. https://doi.org/10.1001/jamaneurol.2020.1127
Butowt R, Bilinska K (2020) SARS-CoV-2: olfaction, brain infection, and the urgent need for clinical samples allowing earlier virus detection. ACS Chem Neurosci 11(9):1200–1203. https://doi.org/10.1021/acschemneuro.0c00172
Zou X, Chen K, Zou J, Han P, Hao J, Han Z (2020) Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med 14(2):185–192. https://doi.org/10.1007/s11684-020-0754-0
Krolewski RC, Packard A, Schwob JE (2013) Global expression profiling of globose basal cells and neurogenic progression within the olfactory epithelium. J Comp Neurol 521(4):833–859. https://doi.org/10.1002/cne.23204
Heydel JM, Coelho A, Thiebaud N, Legendre A, Le Bon AM, Faure P et al (2013) Odorant-binding proteins and xenobiotic metabolizing enzymes: implications in olfactory perireceptor events. Anatomical Record (Hoboken, NJ: 2007) 296(9):1333–1345. https://doi.org/10.1002/ar.22735
Mollica V, Rizzo A, Massari F (2020) The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future oncology (London, England). 16(27):2029–2033. https://doi.org/10.2217/fon-2020-0571
Thunders M, Delahunt B (2020) Gene of the month: TMPRSS2 (transmembrane serine protease 2). J Clin Pathol 73(12):773–776. https://doi.org/10.1136/jclinpath-2020-206987
Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC et al (2001) Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Can Res 61(4):1686–1692
Zmora P, Moldenhauer AS, Hofmann-Winkler H, Pöhlmann S (2015) TMPRSS2 isoform 1 activates respiratory viruses and is expressed in viral target cells. PLoS ONE 10(9):e0138380. https://doi.org/10.1371/journal.pone.0138380
Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I et al (2013) TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87(11):6150–6160. https://doi.org/10.1128/jvi.03372-12
Chen YW, Lee MS, Lucht A, Chou FP, Huang W, Havighurst TC et al (2010) TMPRSS2, a serine protease expressed in the prostate on the apical surface of luminal epithelial cells and released into semen in prostasomes, is misregulated in prostate cancer cells. Am J Pathol 176(6):2986–2996. https://doi.org/10.2353/ajpath.2010.090665
Wettstein L, Kirchhoff F, Münch J (2022) The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. Int J Mol Sci. https://doi.org/10.3390/ijms23031351
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z et al (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181(4):894-904.e9. https://doi.org/10.1016/j.cell.2020.03.045
Lei C, Qian K, Li T, Zhang S, Fu W, Ding M et al (2020) Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun 11(1):2070. https://doi.org/10.1038/s41467-020-16048-4
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (New York, NY). 367(6483):1260–1263. https://doi.org/10.1126/science.abb2507
Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Receptor recognition by the novel Coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS Coronavirus. J Virol. https://doi.org/10.1128/jvi.00127-20
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26(4):450–452. https://doi.org/10.1038/s41591-020-0820-9
Michel CJ, Mayer C, Poch O, Thompson JD (2020) Characterization of accessory genes in coronavirus genomes. Virol J 17(1):131. https://doi.org/10.1186/s12985-020-01402-1
Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I et al (2020) Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci USA 117(13):7001–7003. https://doi.org/10.1073/pnas.2002589117
Hoffmann M, Kleine-Weber H, Pöhlmann S (2020) A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell 78(4):779–84.e5. https://doi.org/10.1016/j.molcel.2020.04.022
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 82(15):7264–7275. https://doi.org/10.1128/jvi.00737-08
Strotmann J, Breer H (2011) Internalization of odorant-binding proteins into the mouse olfactory epithelium. Histochem Cell Biol 136(3):357–369. https://doi.org/10.1007/s00418-011-0850-y
Abbasi AZ, Kiyani DA, Hamid SM, Saalim M, Fahim A, Jalal N (2021) Spiking dependence of SARS-CoV-2 pathogenicity on TMPRSS2. J Med Virol 93(7):4205–4218. https://doi.org/10.1002/jmv.26911
Zhao H, Lu L, Peng Z, Chen LL, Meng X, Zhang C et al (2022) SARS-CoV-2 Omicron variant shows less efficient replication and fusion activity when compared with Delta variant in TMPRSS2-expressed cells. Emerg microb Infect 11(1):277–283. https://doi.org/10.1080/22221751.2021.2023329
Jackson CB, Farzan M, Chen B, Choe H (2022) Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol 23(1):3–20. https://doi.org/10.1038/s41580-021-00418-x
Saito A, Irie T, Suzuki R, Maemura T, Nasser H, Uriu K et al (2022) Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 602(7896):300–306. https://doi.org/10.1038/s41586-021-04266-9
Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC et al (2021) The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol 6(7):899–909. https://doi.org/10.1038/s41564-021-00908-w
Arora P, Sidarovich A, Krüger N, Kempf A, Nehlmeier I, Graichen L et al (2021) B.1.617.2 enters and fuses lung cells with increased efficiency and evades antibodies induced by infection and vaccination. Cell Rep 37(2):109825. https://doi.org/10.1016/j.celrep.2021.109825
Zhang J, Xiao T, Cai Y, Lavine CL, Peng H, Zhu H et al (2021) Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science (New York, NY). 374(6573):1353–1360. https://doi.org/10.1126/science.abl9463
Braga L, Ali H, Secco I, Chiavacci E, Neves G, Goldhill D et al (2021) Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 594(7861):88–93. https://doi.org/10.1038/s41586-021-03491-6
Khan H, Winstone H, Jimenez-Guardeño JM, Graham C, Doores KJ, Goujon C et al (2021) TMPRSS2 promotes SARS-CoV-2 evasion from NCOA7-mediated restriction. PLoS Pathog 17(11):e1009820. https://doi.org/10.1371/journal.ppat.1009820
Hoffmann M, Mösbauer K, Hofmann-Winkler H, Kaul A, Kleine-Weber H, Krüger N et al (2020) Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 585(7826):588–590. https://doi.org/10.1038/s41586-020-2575-3
SeyedAlinaghi S, Mehrtak M, MohsseniPour M, Mirzapour P, Barzegary A, Habibi P et al (2021) Genetic susceptibility of COVID-19: a systematic review of current evidence. Eur J Med Res 26(1):46. https://doi.org/10.1186/s40001-021-00516-8
Ejaz H, Alsrhani A, Zafar A, Javed H, Junaid K, Abdalla AE et al (2020) COVID-19 and comorbidities: deleterious impact on infected patients. J Infect Public Health 13(12):1833–1839. https://doi.org/10.1016/j.jiph.2020.07.014
Sanyaolu A, Okorie C, Marinkovic A, Patidar R, Younis K, Desai P et al (2020) Comorbidity and its Impact on Patients with COVID-19. SN Comp Clin Med 2(8):1069–1076. https://doi.org/10.1007/s42399-020-00363-4
Muschitz C, Trummert A, Berent T, Laimer N, Knoblich L, Bodlaj G et al (2021) Attenuation of COVID-19-induced cytokine storm in a young male patient with severe respiratory and neurological symptoms. Wien Klin Wochenschr 133(17–18):973–978. https://doi.org/10.1007/s00508-021-01867-2
Saengsiwaritt W, Jittikoon J, Chaikledkaew U, Udomsinprasert W (2022) Genetic polymorphisms of ACE1, ACE2, and TMPRSS2 associated with COVID-19 severity: a systematic review with meta-analysis. Rev Med Virol. https://doi.org/10.1002/rmv.2323
Pandey RK, Srivastava A, Singh PP, Chaubey G (2022) Genetic association of TMPRSS2 rs2070788 polymorphism with COVID-19 case fatality rate among Indian populations. Infect Genet Evol 98:105206. https://doi.org/10.1016/j.meegid.2022.105206
Paniri A, Hosseini MM, Akhavan-Niaki H (2021) First comprehensive computational analysis of functional consequences of TMPRSS2 SNPs in susceptibility to SARS-CoV-2 among different populations. J Biomol Struct Dyn 39(10):3576–3593. https://doi.org/10.1080/07391102.2020.1767690
Bhanushali A, Rao P, Raman V, Kokate P, Ambekar A, Mandva S et al (2018) Status of TMPRSS2-ERG fusion in prostate cancer patients from India: correlation with clinico-pathological details and TMPRSS2 Met160Val polymorphism. Prostate Int 6(4):145–150. https://doi.org/10.1016/j.prnil.2018.03.004
Maekawa S, Suzuki M, Arai T, Suzuki M, Kato M, Morikawa T et al (2014) TMPRSS2 Met160Val polymorphism: significant association with sporadic prostate cancer, but not with latent prostate cancer in Japanese men. Int J Urol 21(12):1234–1238. https://doi.org/10.1111/iju.12578
Giri VN, Ruth K, Hughes L, Uzzo RG, Chen DY, Boorjian SA et al (2011) Racial differences in prediction of time to prostate cancer diagnosis in a prospective screening cohort of high-risk men: effect of TMPRSS2 Met160Val. BJU Int 107(3):466–470. https://doi.org/10.1111/j.1464-410X.2010.09522.x
Lubieniecka JM, Cheteri MK, Stanford JL, Ostrander EA (2004) Met160Val polymorphism in the TRMPSS2 gene and risk of prostate cancer in a population-based case-control study. Prostate 59(4):357–359. https://doi.org/10.1002/pros.20005
Wulandari L, Hamidah B, Pakpahan C, Damayanti NS, Kurniati ND, Adiatmaja CO et al (2021) Initial study on TMPRSS2 p.Val160Met genetic variant in COVID-19 patients. Human Genom 15(1):29. https://doi.org/10.1186/s40246-021-00330-7
Karimian M, Aftabi Y, Mazoochi T, Babaei F, Khamechian T, Boojari H et al (2018) Survivin polymorphisms and susceptibility to prostate cancer: a genetic association study and an in silico analysis. EXCLI J 17:479–491
Mobasseri N, Babaei F, Karimian M, Nikzad H (2018) Androgen receptor (AR)-CAG trinucleotide repeat length and idiopathic male infertility: a case-control trial and a meta-analysis. EXCLI J 17:1167–1179
Bafrani HH, Ahmadi M, Jahantigh D, Karimian M (2019) Association analysis of the common varieties of IL17A and IL17F genes with the risk of knee osteoarthritis. J Cell Biochem 120(10):18020–18030
Mobasseri N, Nikzad H, Karimian M (2019) Protective effect of oestrogen receptor α-PvuII transition against idiopathic male infertility: a case-control study and meta-analysis. Reprod Biomed Online 38(4):588–598
Karimian M, Momeni A, Farmohammadi A, Behjati M, Jafari M, Raygan F (2020) Common gene polymorphism in ATP-binding cassette transporter A1 and coronary artery disease: a genetic association study and a structural analysis. J Cell Biochem 121(5–6):3345–3357
Karimian M, Behjati M, Barati E, Ehteram T, Karimian A (2020) CYP1A1 and GSTs common gene variations and presbycusis risk: a genetic association analysis and a bioinformatics approach. Environ Sci Pollut Res 27(34):42600–42610
Karimian M, Ghazaey Zidanloo S, Jahantigh D (2022) Influence of FOXP3 gene polymorphisms on the risk of preeclampsia: a meta-analysis and a bioinformatic approach. Clin Exp Hypertens 44(3):280–290
Zamani-Badi T, Karimian M, Azami-Tameh A, Nikzad H (2019) Association of C3953T transition in interleukin 1β gene with idiopathic male infertility in an Iranian population. Hum Fertil 22:111–117
Karimian M, Hosseinzadeh CA (2018) Human MTHFR-G1793A transition may be a protective mutation against male infertility: a genetic association study and in silico analysis. Hum Fertil 21(2):128–136
Torre-Fuentes L, Matías-Guiu J, Hernández-Lorenzo L, Montero-Escribano P, Pytel V, Porta-Etessam J et al (2021) ACE2, TMPRSS2, and Furin variants and SARS-CoV-2 infection in Madrid, Spain. J Med Virol 93(2):863–869. https://doi.org/10.1002/jmv.26319
Monticelli M, Hay Mele B, Benetti E, Fallerini C, Baldassarri M, Furini S et al (2021) Protective role of a TMPRSS2 variant on severe COVID-19 outcome in young males and elderly women. Genes. https://doi.org/10.3390/genes12040596
Curtis D (2020) Variants in ACE2 and TMPRSS2 genes are not major determinants of COVID-19 severity in UK Biobank Subjects. Hum Hered 85(2):66–68. https://doi.org/10.1159/000515200
Schönfelder K, Breuckmann K, Elsner C, Dittmer U, Fistera D, Herbstreit F et al (2021) Transmembrane serine protease 2 polymorphisms and susceptibility to severe acute respiratory syndrome Coronavirus Type 2 Infection: a German case-control study. Front Genet 12:667231. https://doi.org/10.3389/fgene.2021.667231
Ravikanth V, Sasikala M, Naveen V, Latha SS, Parsa KVL, Vijayasarathy K et al (2021) A variant in TMPRSS2 is associated with decreased disease severity in COVID-19. Meta Gene 29:100930. https://doi.org/10.1016/j.mgene.2021.100930
Akin S, Schriek P, van Nieuwkoop C, Neuman RI, Meynaar I, van Helden EJ et al (2022) A low aldosterone/renin ratio and high soluble ACE2 associate with COVID-19 severity. J Hypertens 40(3):606–614. https://doi.org/10.1097/hjh.0000000000003054
Báez-Santos YM, St John SE, Mesecar AD (2015) The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115:21–38. https://doi.org/10.1016/j.antiviral.2014.12.015
Shen LW, Qian MQ, Yu K, Narva S, Yu F, Wu YL et al (2020) Inhibition of Influenza A virus propagation by benzoselenoxanthenes stabilizing TMPRSS2 Gene G-quadruplex and hence down-regulating TMPRSS2 expression. Sci Rep 10(1):7635. https://doi.org/10.1038/s41598-020-64368-8
Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R Jr, Nunneley JW et al (2015) Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 116:76–84. https://doi.org/10.1016/j.antiviral.2015.01.011
Yamaya M, Shimotai Y, Hatachi Y, Lusamba Kalonji N, Tando Y, Kitajima Y et al (2015) The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm Pharmacol Ther 33:66–74. https://doi.org/10.1016/j.pupt.2015.07.001
Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F (2010) Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84(24):12658–12664. https://doi.org/10.1128/jvi.01542-10
Glowacka I, Bertram S, Müller MA, Allen P, Soilleux E, Pfefferle S et al (2011) Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol 85(9):4122–4134. https://doi.org/10.1128/jvi.02232-10
Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S (2012) Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol 86(12):6537–6545. https://doi.org/10.1128/jvi.00094-12
Ikeda S, Manabe M, Muramatsu T, Takamori K, Ogawa H (1988) Protease inhibitor therapy for recessive dystrophic epidermolysis bullosa. In vitro effect and clinical trial with camostat mesylate. J Am Acad Dermatol 18(6):1246–1252. https://doi.org/10.1016/s0190-9622(88)70130-9
Ohkoshi M, Fujii S (1983) Effect of the synthetic protease inhibitor [N, N-dimethylcarbamoyl-methyl 4-(4-guanidinobenzoyloxy)-phenylacetate] methanesulfate on carcinogenesis by 3-methylcholanthrene in mouse skin. J Natl Cancer Inst 71(5):1053–1057
Ohkoshi M, Oka T (1984) Clinical experience with a protease inhibitor [N, N-dimethylcarbamoylmethyl 4-(4-guanidinobenzoyloxy)-phenylacetate] methanesulfate for prevention of recurrence of carcinoma of the mouth and in treatment of terminal carcinoma. J Maxillofac Surg 12(4):148–152. https://doi.org/10.1016/s0301-0503(84)80235-0
Sai JK, Suyama M, Kubokawa Y, Matsumura Y, Inami K, Watanabe S (2010) Efficacy of camostat mesilate against dyspepsia associated with non-alcoholic mild pancreatic disease. J Gastroenterol 45(3):335–341. https://doi.org/10.1007/s00535-009-0148-1
Yamawaki H, Futagami S, Kaneko K, Agawa S, Higuchi K, Murakami M et al (2019) Camostat mesilate, pancrelipase, and rabeprazole combination therapy improves epigastric pain in early chronic pancreatitis and functional dyspepsia with pancreatic enzyme abnormalities. Digestion 99(4):283–292. https://doi.org/10.1159/000492813
Ramsey ML, Nuttall J, Hart PA (2019) A phase 1/2 trial to evaluate the pharmacokinetics, safety, and efficacy of NI-03 in patients with chronic pancreatitis: study protocol for a randomized controlled trial on the assessment of camostat treatment in chronic pancreatitis (TACTIC). Trials 20(1):501. https://doi.org/10.1186/s13063-019-3606-y
Göke B, Stöckmann F, Müller R, Lankisch PG, Creutzfeldt W (1984) Effect of a specific serine protease inhibitor on the rat pancreas: systemic administration of camostate and exocrine pancreatic secretion. Digestion 30(3):171–178. https://doi.org/10.1159/000199102
Adler G, Müllenhoff A, Koop I, Bozkurt T, Göke B, Beglinger C et al (1988) Stimulation of pancreatic secretion in man by a protease inhibitor (camostate). Eur J Clin Invest 18(1):98–104. https://doi.org/10.1111/j.1365-2362.1988.tb01173.x
Iwaki M, Ino Y, Motoyoshi A, Ozeki M, Sato T, Kurumi M et al (1986) Pharmacological studies of FUT-175, nafamostat mesilate. V. Effects on the pancreatic enzymes and experimental acute pancreatitis in rats. Jpn J Pharmacol 41(2):155–162. https://doi.org/10.1254/jjp.41.155
Hiraishi M, Yamazaki Z, Ichikawa K, Kanai F, Idezuki Y, Onishi K et al (1988) Plasma collection using nafamostat mesilate and dipyridamole as an anticoagulant. Int J Artif Organs 11(3):212–216
Hirota M, Shimosegawa T, Kitamura K, Takeda K, Takeyama Y, Mayumi T et al (2020) Continuous regional arterial infusion versus intravenous administration of the protease inhibitor nafamostat mesilate for predicted severe acute pancreatitis: a multicenter, randomized, open-label, phase 2 trial. J Gastroenterol 55(3):342–352. https://doi.org/10.1007/s00535-019-01644-z
Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue JI et al (2016) Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome Coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother 60(11):6532–6539. https://doi.org/10.1128/aac.01043-16
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England) 395(10224):565–574. https://doi.org/10.1016/s0140-6736(20)30251-8
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181(2):281–92.e6. https://doi.org/10.1016/j.cell.2020.02.058
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M et al (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30(3):269–271. https://doi.org/10.1038/s41422-020-0282-0
Funding
This research did not receive any specific grant.
Author information
Authors and Affiliations
Contributions
MK participated in the design of the study. AK and MB participated in searching for content in reputable research databases and categorizing content. AK and MB participated in providing an initial draft of the manuscript. MK participated in editing and organizing the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
None of the authors declare a conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Pablo Pineyro.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Karimian, A., Behjati, M. & Karimian, M. Molecular mechanisms involved in anosmia induced by SARS-CoV-2, with a focus on the transmembrane serine protease TMPRSS2. Arch Virol 167, 1931–1946 (2022). https://doi.org/10.1007/s00705-022-05545-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05545-0