
Abstract
Smacoviruses have small (∼2.3-2.9 kb), circular single-stranded DNA genomes encoding rolling circle replication-associated proteins (Rep) and unique capsid proteins. Although smacoviruses are prevalent in faecal matter of various vertebrates, including humans, none of these viruses have been cultured thus far. Smacoviruses display ∼45% genome-wide sequence diversity, which is very similar to that found within other families of circular Rep-encoding single-stranded (CRESS) DNA viruses, including members of the families Geminiviridae (46% diversity) and Genomoviridae (47% diversity). Here, we announce the creation of a new family Smacoviridae and describe a sequence-based taxonomic framework which was used to classify 83 smacovirus genomes into 43 species within six new genera, Bovismacovirus (n=3), Cosmacovirus (n=1), Dragsmacovirus (n=1), Drosmacovirus (n=3), Huchismacovirus (n=7), and Porprismacovirus (n=28). As in the case of genomoviruses, the species demarcation is based on the genome-wide pairwise identity, whereas genera are established based on the Rep amino acid sequence identity coupled with strong phylogenetic support. A similar sequence-based taxonomic framework should guide the classification of an astonishing diversity of other uncultured and currently unclassified CRESS DNA viruses discovered by metagenomic approaches.
Similar content being viewed by others

With the advent of metagenomics approaches, a large diversity of unknown viruses has been uncovered in various environmental, plant, and animal samples [23]. Sampling of animal faecal matter has proved to be particularly efficient for the discovery of a wide variety of novel viral types, in particular those with small DNA genomes. Until recently, the circular replication-initiation protein encoding single-stranded (CRESS) DNA viruses associated with eukaryotic hosts have been classified by the International Committee on Taxonomy of Viruses (ICTV) into four families, namely Circoviridae, Genomoviridae, Geminiviridae and Nanoviridae. In 2018, the ICTV created two new families for classification of CRESS DNA viruses, Bacilladnaviridae and Smacoviridae. The family Bacilladnaviridae, which includes viruses infecting diatoms, a major group of unicellular algae widespread in aquatic habitats, has been described elsewhere [7]. Here, we introduce the family Smacoviridae (smaco- stands for small circular DNA viruses) and describe the ICTV-approved sequence-based taxonomic framework for classification of these viruses.
Smacoviruses [15, 17], previously also referred to as chipoviruses [22, 24], have been identified in faecal matter of various vertebrates, including humans, as well as in the abdomina of dragonflies of two species (Table 1). Thus far, none of these viruses have been cultured or found in animal tissue sample. Nonetheless, the viruses have been discovered using viral metagenomics approaches and for the majority of the smacoviruses, the validity of genome sequences has been verified by either PCR amplification using abutting primers followed by Sanger sequencing of these products or by amplification, cloning and Sanger sequencing of the recombinant plasmids [1, 3, 4, 9, 15, 22, 24].
The genomes of currently identified smacoviruses are ~2300-2900 nucleotides-long, contain two major open reading frames (ORF), encoding the rolling circle replication-associated protein (Rep) and capsid protein (CP; Figure 1). The two ORFs in all 83 smacovirus genomes are bidirectionally organised, separated by two intergenic regions. Similar to other CRESS DNA viruses, smacoviruses contain a conserved nonanucleotide sequence located at a putative stem-loop structure at the origin of replication (Figure 1), where nicking of the dsDNA replicative intermediate is predicted to occur. The Reps of smacoviruses are homologous but phylogenetically distinct from those of classified CRESS DNA viruses (Figure 2). Phylogenetic analysis and comparison of the conserved sequence motifs suggest a closer evolutionary relationship between the smacovirus and nanovirus Reps [7]. By contrast, although conserved among smacoviruses, the CPs do not display recognizable sequence similarity to the CPs of other known viruses.

Genome organization of a representative smacovirus (chimpanzee associated porprismacovirus 1 [GQ351272]) and a WebLogo of the nonanucleotide motif found in smacoviruses

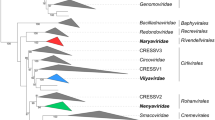
Unrooted approximate maximum likelihood phylogenetic tree of Reps of CRESS DNA viruses inferred using FastTree [18]. Major groups of classified CRESS DNA viruses as well as alphasatellites associated with geminiviruses and nanoviruses are colour coded
Analysis of the genome-wide pairwise identities of the 83 smacoviruses (Figure 3) shows 45% diversity amongst these genomes, which is similar to values determined for members of the families Geminiviridae [27] and Genomoviridae [12, 25]. The plot of the distribution of pairwise identities shows a trough between 76 and 88%. Hence, for this group of viruses, 77% genome-wide pairwise identity is chosen as a species demarcation threshold. Using this approach, the 83 smacoviruses were assigned to 43 species (Table 1).

Distribution of pairwise identities of the full genome (upper panel), the replication initiation protein (middle panel) and the capsid protein sequences determined using SDT v1.2 [14]. The arrows indicate the thresholds of the full genome (top panel) and Rep protein (middle panel) pairwise sequence identities used as the species and genus demarcation criteria, respectively
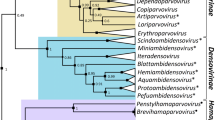
Maximum likelihood phylogenetic analysis of the Rep sequences of all 83 smacoviruses reveals four main clusters with >90% branch support and two singletons (Figure 4). Rep sequences within each of the four clades in general share >40% pairwise identity, whereas sequences from different phylogenetic clades show less than 40% identity to each other. We note that phylogenetic trees produced using complete genome (Figure 5) and CP (Figure 6) sequences are not congruent with the Rep phylogeny, presumably due to intra-familial recombination between different smacovirus genomes resulting in chimeric entities encoding Rep and CP with different evolutionary histories, as has been also observed for genomoviruses [25]. Given that smacovirus Reps are considerably more conserved than CPs (Figure 3) and due to the fact that Reps are the only proteins shared across all CRESS DNA viruses [11, 20], genera were established based on the phylogenetic analysis of the Rep sequences coupled with their pairwise sequence identity. Accordingly, 40% Rep amino acid sequence identity coupled with strong phylogenetic support is proposed as a genus level demarcation threshold.

Maximum likelihood phylogenetic tree of the Rep amino acid sequences of the smacoviruses inferred using PhyML [6] with the LG+G+I+F substitution model. The tree is rooted with the Rep sequences of nanoviruses

Maximum likelihood phylogenetic tree of the genome sequences inferred using IQ-TREE [16] with K3Pu+I+G4 substitution model. Branches with <60% bootstrap support have been collapsed and the tree is mid-point rooted

Maximum likelihood phylogenetic tree of the CP amino acid sequences of the smacoviruses inferred using PhyML [6] with the LG+G+I+F substitution model. The phylogenetic tree is mid-point rooted
The naming practice for smacoviruses and other uncultivated CRESS DNA viruses, such as genomoviruses [25], typically involves adoption of the name of an organism or material from which the virus genome has been sequenced. In the absence of evidence of actual infection, the word “associated” is usually added to the potential host name to emphasize that the organism may or may not be the actual host. As a case in point, it has been recently suggested that dsRNA viruses of the family Picobirnaviridae, which for three decades were considered to infect eukaryotes [5], might instead replicate in bacteria that populate the enteric tract of animals [10]. Thus, utmost caution should be exercised when assigning viruses to potential hosts.
The following names for the six genera within the Smacoviridae have been adopted
Bovismacovirus: Bovine smacovirus
3 species (Table 1);
Drosmacovirus: Dromedary smacovirus
3 species (Table 1);
Huchismacovirus: Human and chicken smacovirus
7 species (Table 1);
Porprismacovirus: Porcine and primate smacovirus
28 species (Table 1);
Cosmacovirus: Cow smacovirus
1 species (Table 1);
Dragsmacovirus: Dragonfly smacovirus.
1 species (Table 1).
We would like to note that the species Sheep associated porprismacovirus 3, Bovine associated huchismacovirus 1 and Bovine associated huchismacovirus 2 have been tentatively assigned to genera Porprismacovirus and Huchismacovirus. It is highly likely that, as more sequences become available, new genera will have to be created for these divergent smacoviruses (Figures 4 and 5).
Sequence based taxonomic framework employed here for smacoviruses and previously applied for genomoviruses [25] should guide the classification of an astonishing diversity of other uncultured CRESS DNA viruses described by metagenomic approaches.
Change history
27 July 2018
Unfortunately Fig. 6 in Archives of Virology (2018) 163:2005–2015 https://doi.org/10.1007/s00705-018-3820-z is duplicated (Fig. 4). This is corrected in this erratum.
References
Blinkova O, Victoria J, Li Y, Keele BF, Sanz C, Ndjango JB, Peeters M, Travis D, Lonsdorf EV, Wilson ML, Pusey AE, Hahn BH, Delwart EL (2010) Novel circular DNA viruses in stool samples of wild-living chimpanzees. J Gen Virol 91:74–86
Cheung AK, Ng TF, Lager KM, Bayles DO, Alt DP, Delwart EL, Pogranichniy RM, Kehrli ME (2013) A divergent clade of circular single-stranded DNA viruses from pig feces. Adv Virol 158:2157–2162
Cheung AK, Ng TFF, Lager KM, Alt DP, Delwart E, Pogranichniy RM (2015) Identification of several clades of novel single-stranded circular DNA viruses with conserved stem-loop structures in pig feces. Adv Virol 160:353–358
Dayaram A, Potter KA, Pailes R, Marinov M, Rosenstein DD, Varsani A (2015) Identification of diverse circular single-stranded DNA viruses in adult dragonflies and damselflies (Insecta: Odonata) of Arizona and Oklahoma, USA. Infect Genet Evol 30:278–287
Ganesh B, Masachessi G, Mladenova Z (2014) Animal picobirnavirus. Virusdisease 25:223–238
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Kazlauskas D, Dayaram A, Kraberger S, Goldstien S, Varsani A, Krupovic M (2017) Evolutionary history of ssDNA bacilladnaviruses features horizontal acquisition of the capsid gene from ssRNA nodaviruses. Virology 504:114–121
Kim AR, Chung HC, Kim HK, Kim EO, Nguyen VG, Choi MG, Yang HJ, Kim JA, Park BK (2014) Characterization of a complete genome of a circular single-stranded DNA virus from porcine stools in Korea. Virus Genes 48:81–88
Kim HK, Park SJ, Nguyen VG, Song DS, Moon HJ, Kang BK, Park BK (2012) Identification of a novel single-stranded, circular DNA virus from bovine stool. J Gen Virol 93:635–639
Krishnamurthy SR, Wang D (2018) Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 516:108–114
Krupovic M (2013) Networks of evolutionary interactions underlying the polyphyletic origin of ssDNA viruses. Curr Opin Virol 3:578–586
Krupovic M, Ghabrial SA, Jiang D, Varsani A (2016) Genomoviridae: a new family of widespread single-stranded DNA viruses. Arch Virol 161:2633–2643
Lima DA, Cibulski SP, Finkler F, Teixeira TF, Varela APM, Cerva C, Loiko MR, Scheffer CM, Dos Santos HF, Mayer FQ, Roehe PM (2017) Faecal virome of healthy chickens reveals a large diversity of the eukaryote viral community, including novel circular ssDNA viruses. J Gen Virol 98:690–703
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9:e108277
Ng TFF, Zhang W, Sachsenröder J, Kondov NO, da Costa AC, Vega E, Holtz LR, Wu G, Wang D, Stine CO, Antonio M, Mulvaney US, Muench MO, Deng X, Ambert-Balay K, Pothier P, Vinjé J, Delwart E (2015) A diverse group of small circular ssDNA viral genomes in human and non-human primate stools. Virus Evol 1:vev017
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Phan TG, da Costa AC, Del Valle Mendoza J, Bucardo-Rivera F, Nordgren J, O’Ryan M, Deng X, Delwart E (2016) The fecal virome of South and Central American children with diarrhea includes small circular DNA viral genomes of unknown origin. Arch Virol 161:959–966
Price MN, Dehal PS, Arkin AP (2010) FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490
Reuter G, Boros A, Delwart E, Pankovics P (2014) Novel circular single-stranded DNA virus from turkey faeces. Adv Virol 159:2161–2164
Rosario K, Duffy S, Breitbart M (2012) A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Arch Virol 157:1851–1871
Sachsenroder J, Braun A, Machnowska P, Ng TF, Deng X, Guenther S, Bernstein S, Ulrich RG, Delwart E, Johne R (2014) Metagenomic identification of novel enteric viruses in urban wild rats and genome characterization of a group A rotavirus. J Gen Virol 95:2734–2747
Sikorski A, Arguello-Astorga GR, Dayaram A, Dobson RC, Varsani A (2013) Discovery of a novel circular single-stranded DNA virus from porcine faeces. Arch Virol 158:283–289
Simmonds P, Adams MJ, Benko M, Breitbart M, Brister JR, Carstens EB, Davison AJ, Delwart E, Gorbalenya AE, Harrach B, Hull R, King AM, Koonin EV, Krupovic M, Kuhn JH, Lefkowitz EJ, Nibert ML, Orton R, Roossinck MJ, Sabanadzovic S, Sullivan MB, Suttle CA, Tesh RB, van der Vlugt RA, Varsani A, Zerbini FM (2017) Consensus statement: virus taxonomy in the age of metagenomics. Nat Rev Microbiol 15:161–168
Steel O, Kraberger S, Sikorski A, Young LM, Catchpole RJ, Stevens AJ, Ladley JJ, Coray DS, Stainton D, Dayarama A, Julian L, van Bysterveldt K, Varsani A (2016) Circular replication-associated protein encoding DNA viruses identified in the faecal matter of various animals in New Zealand. Infect Genet Evol 43:151–164
Varsani A, Krupovic M (2017) Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol 3:vew037
Woo PC, Lau SK, Teng JL, Tsang AK, Joseph M, Wong EY, Tang Y, Sivakumar S, Bai R, Wernery R, Wernery U, Yuen KY (2014) Metagenomic analysis of viromes of dromedary camel fecal samples reveals large number and high diversity of circoviruses and picobirnaviruses. Virology 471–473:117–125
Zerbini FM, Briddon RW, Idris A, Martin DP, Moriones E, Navas-Castillo J, Rivera-Bustamante R, Roumagnac P, Varsani A, Ictv Report C (2017) ICTV virus taxonomy profile: Geminiviridae. J Gen Virol 98:131–133
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare there are no conflicts of interest.
Research involving human participants and/or animals
The research did not involve human participants or animals.
Informed consent
The research did not involve human participants or animals.
Additional information
Handling Editor: Sead Sabanadzovic.
Rights and permissions
About this article

Cite this article
Varsani, A., Krupovic, M. Smacoviridae: a new family of animal-associated single-stranded DNA viruses. Arch Virol 163, 2005–2015 (2018). https://doi.org/10.1007/s00705-018-3820-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-3820-z