
Abstract
A novel bipartite legumovirus (genus Begomovirus, family Geminiviridae), that naturally infects the wild leguminous plant Desmodium sp. in Uganda, was molecularly characterized and named Desmodium mottle virus. The highest nucleotide identities for DNA-A, obtained from two field-collected samples, were 79.9% and 80.1% with the legumovirus, soybean mild mottle virus. DNA-B had the highest nucleotide identities (65.4% and 66.4%) with a typical non-legumovirus Old World begomovirus, African cassava mosaic virus. This is the first report of a legumovirus in East Africa and extends the known diversity of begomoviruses found infecting wild plants in this continent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The family Geminiviridae comprises seven genera, differentiated based on genome organization, nucleotide sequence identity and biological properties: Begomovirus, Mastrevirus, Eragrovirus, Curtovirus, Turncurtovirus, Topocuvirus and Becurtovirus [4, 27]. The genus Begomovirus is the largest in the family, with 322 accepted species [5, https://talk.ictvonline.org/ictv_wikis/geminiviridae/m/files_gemini/5120]. Begomoviruses are transmitted by the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) and frequently cause important plant-diseases around the world [21]. Bipartite begomoviruses possess two genome components (DNA-A and DNA-B), which are essential for virus infectivity and the size of each component ranges between 2.5 and 2.8 kb. DNA-A and DNA-B share ~200 nt in the common region (CR), located within the intergenic region, that contains cis elements for replication and control of gene expression. The CR exhibits a high degree of sequence identity between both genome components of bipartite begomoviruses [4]. Based on the phylogenetic analysis of complete nucleotide sequences of DNA-A, begomoviruses can be classified into four lineages, Old World (OW), New World (NW), sweepoviruses and legumoviruses [3].
Legumoviruses, or legume-infecting begomoviruses from the OW, are amongst the most atypical begomoviruses [16]. They are distinct from the numerous legume-infecting begomoviruses that occur in the Americas and in phylogenetic analyses they group in a cluster different from those of OW and NW begomoviruses [3, 10]. The difference between legumoviruses and typical OW begomoviruses could have arisen due to genetic isolation involving either a host-range barrier or lack of movement of whitefly vectors between legumes and non-leguminous plants, thereby preventing genetic exchange between both groups of viruses [24]. The genomes of most legumoviruses are bipartite, although a DNA-B component has not been identified for cowpea golden mosaic virus (CPGMV), Dolichos yellow mosaic virus (DoYMV) and soybean mild mottle virus (SbMMoV) [1, 19]. Little attention has been paid to legumoviruses infecting wild plants. Scarce examples include DoYMV infecting Lablab purpureus (sin. Dolichos lablab) [19] and horsegram yellow mosaic virus (HgYMV) infecting Macrotyloma uniflorum [2] in India, kudzu mosaic virus (KuMV) infecting Pueraria montana in Vietnam [13], soybean chlorotic blotch virus (SbCBV) infecting Centrosema pubescens in Nigeria [1] and Rhynchosia yellow mosaic virus (RhYMV) infecting Rhynchosia minima in Pakistan [15].
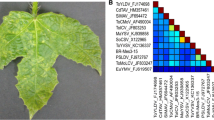
In this study, leaf samples of two Desmodium sp. (family Leguminosae) plants showing mottle symptoms (Fig. 1) were collected in Kikonge, southwestern Uganda, in March 2015 (00°22.641’ N; 32°11.252’ E [sample UG4], 00°22.640’ N; 32°11.252’ E [sample UG5]). Morphological identification of the plant samples at the genus level was confirmed molecularly by DNA barcoding using chloroplast rbcL and matK genes [14]. The genus Desmodium is composed of about 370 accepted species native to tropical East Asia, Africa and America. Some Desmodium species are considered as weeds, although others containing potent secondary metabolites are used in agriculture in push-pull technology [7].

Desmodium sp. plants analyzed in this work showing mottle symptoms on leaves. (A) sample UG4, (B) sample UG5
Total nucleic acids were extracted from leaf samples using a modified CTAB method [22]. To test for the presence of begomoviruses in the samples, putatively causing the observed symptoms, nucleic acids were used as a template for rolling-circle amplification (RCA) using φ29 DNA polymerase (TempliPhi kit, GE Healthcare) and amplified RCA products were digested with a set of restriction enzymes (ClaI, BamHI, EcoRI, HindIII, NcoI, NheI and SalI) [17]. Putative full length begomoviral genomic components (~2.8 kbp) were cloned from each sample (EcoRI and HindIII for UG4 and ClaI and EcoRI for UG5) into pBlueScript II SK (+) (Stratagene) and selected clones (one per sample and restriction enzyme) were sequenced at Macrogen Inc. (Seoul, South Korea). Sequences were assembled with SeqMan software included in the DNASTAR package (DNASTAR Inc.). Open reading frames (ORFs) were identified using Open Reading Frame Finder (NCBI) and confirmed using the BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi) on their deduced amino acid sequences. Initial sequence identity comparison was performed using the BLAST program, sequences were aligned with MUSCLE [9] and pairwise identity scores were calculated using SDT (Sequence demarcation tool) [20]. A phylogenetic analysis using maximum likelihood (ML) was used after selecting the best-fit model of nucleotide substitution based on corrected Akaike Information Criterion (AICc) and Bayesian Information Criterion (BIC) as implemented in MEGA 6 [25]. Recombination analysis was performed using RDP4 [18] after alignment, with MUSCLE, of the sequences selected with SWeBLAST (with a window size of 200 and a step size of 200) [12]. SWeBLAST avoids the significant problem of deciding which sequences to compare, thus allowing identification of putative parents of recombinant sequences (four sequences for DNA-A and nine sequences for DNA-B). Only recombination events detected with at least five methods with p-values lower than 10−2 were considered.
The cloned genome components from each sample were shown to correspond to begomoviral DNA-A and DNA-B components. DNA-A component from sample UG4 (2767 nt, EcoRI fragment, KY294724) and UG5 (2767 nt, ClaI fragment, KY294725) showed the highest nucleotide sequence identity (79.9% and 80%, respectively) to SbMMoV (GQ472984), a legumovirus found in soybean (Glycine max) in Nigeria [1]. The DNA-B component from sample UG4 (2715 nt, HindIII fragment, KY294726) exhibited the highest nucleotide sequence identity (65.4%) with an isolate of African cassava mosaic virus (ACMV) (KJ887741) from Madagascar [8], while UG5 DNA-B (2713 nt, EcoRI fragment, KY294727) exhibited the highest nucleotide sequence identity (66.4%) with another isolate of ACMV (HE616778) from Burkina Faso [26]. Pairwise nucleotide identities between DNA-A and DNA-B from samples UG4 and UG5 were 100% and 99.7%, respectively, confirming that the virus identified from both samples belonged to the same begomovirus species. Also, the virus showed a typical genome organization of Old World bipartite begomoviruses. In accordance, therefore, with current taxonomic guidelines for the genus Begomovirus (a new DNA-A sequence with less than 91% pairwise identity to any other published begomovirus DNA-A sequence will belong to a new begomovirus species) [5], the isolates described here ([Uganda-Kikonge UG4-2015] and [Uganda-Kikonge UG5-2015]) represent a novel species for which we propose the name Desmodium mottle virus (DesMoV).
DNA-A and DNA-B from both samples showed a CR of 179 nt (DNA-A) and 156 nt (DNA-B) with sequence identities of 90.2% (sample UG4) and 90.8% (sample UG5). The difference in length of the CR is due to a deletion in DNA-B. Both components from each sample showed three copies of iterons (AATCGGGGGT) (one is inverted and the most proximal to TATA box is imperfect), indicating that DNA-A and DNA-B isolated from each sample constitute a cognate pair.
A phylogenetic tree based on alignment of the DNA-A sequences obtained here with those of selected begomoviruses (including one sequence from each legumovirus species) showed that they grouped in a cluster with the legumoviruses (Fig. 2A). However, DNA-Bs grouped with ACMV, a typical OW begomovirus (Fig. 2B). Similar phylogenetic relationships have been described previously for SbCBV, the only bipartite legumovirus identified in Africa until now [1]. This is an example of the distinct evolutionary history undergone by the DNA-A and DNA-B genome components, as shown previously for other begomoviruses [3, 6, 11, 23, 24]. No recombination event was detected in any of the genome components of DesMoV.

Phylogenetic trees illustrating the relationship of isolates of Desmodium mottle virus (DesMoV) DNA-A (A) and DNA-B (B) to other begomoviruses. The trees were constructed by the maximum-likelihood method (1000 replicates) with the MEGA 6 program using the best fit model, TN93+G+I for DNA-A and HKY+G+I for DNA-B. ACMV, African cassava mosaic virus; AYVV, Ageratum yellow vein virus; CLCuGeV, cotton leaf curl Gezira virus; CMMGV, cassava mosaic Madagascar virus; CPGMV, cowpea golden mosaic virus; DoYMV, Dolichos yellow mosaic virus; EACMV, East African cassava mosaic virus; HgYMV, horsegram yellow mosaic virus; KuMV, kudzu mosaic virus; MYMIV, mungbean yellow mosaic India virus; MYMV, mungbean yellow mosaic virus; PouGMV, Pouzolzia golden mosaic virus; RhYMV, Rhynchosia yellow mosaic virus; SbCBV, soybean chlorotic blotch virus; SbMMoV, soybean mild mottle virus; SgYVV, Siegesbeckia yellow vein virus; ToCSV, tomato curly stunt virus; ToLCPatV, tomato leaf curl Patna virus. A set of New World begomoviruses was used as the outgroup. The bar below each tree indicates nucleotide substitutions per site
DesMoV is the first legumovirus to be described from East Africa to date and because it is phylogenetically closely related to begomoviruses that infect soybean or cowpea in West Africa, it may represent a potential threat to these crops. Additional research work to investigate its host-plant range and whitefly transmission characteristics, therefore, should be initiated to assess the threat to crops posed by this newly discovered begomovirus.
References
Alabi OJ, Kumar PL, Mgbechi-Ezeri JU, Rayapati AN (2010) Two new ‘legumoviruses’ (genus Begomovirus) naturally infecting soybean in Nigeria. Arch Virol 155:643–656
Barnabas AD, Radhakrishnan GK, Usha R (2010) Characterization of a begomovirus causing horsegram yellow mosaic disease in India. Eur J Plant Pathol 127:41–51
Briddon RW, Patil BL, Bagewadi B, Nawaz-ul-Rehman MS, Fauquet CM (2010) Distinct evolutionary histories of the DNA-A and DNA-B components of bipartite begomoviruses. BMC Evol Biol 10:97
Brown JK, Fauquet CM, Briddon RW, Zerbini M, Moriones E, Navas-Castillo J (2012) Family Geminiviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy. Ninth report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press, London, pp 351–373
Brown JK, Zerbini FM, Navas-Castillo J, Moriones E, Ramos-Sobrinho R, Silva JCF, Fiallo-Olivé E, Briddon RW, Hernández-Zepeda C, Idris A, Malathi VG, Martin DP, Rivera-Bustamante R, Ueda S, Varsani A (2015) Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch Virol 160:1593–1619
Bull SE, Briddon RW, Sserubombwe WS, Ngugi K, Markham PG, Stanley J (2006) Genetic diversity and phylogeography of cassava mosaic viruses in Kenya. J Gen Virol 87:3053–3065
Cook SM, Khan ZR, Pickett JA (2007) The use of push-pull strategies in integrated pest management. Annu Rev Entomol 52:375–400
De Bruyn A, Harimalala M, Zinga M, Mabvakure BM, Hoareau M, Ravigné V, Walters M, Reynaud B, Varsani A, Harkins GW, Martin DP, Lett JM, Lefeuvre P (2016) Divergent evolutionary and epidemiological dynamics of cassava mosaic geminiviruses in Madagascar. BMC Evol Biol 16:182
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Fauquet CM, Briddon RW, Brown JK, Moriones E, Stanley J, Zerbini M, Zhou X (2008) Geminivirus strain demarcation and nomenclature. Arch Virol 153:783–821
Fiallo-Olivé E, Navas-Castillo J, Moriones E, Martínez-Zubiaur Y (2010) Two novel begomoviruses belonging to different lineages infecting Rhynchosia minima. Arch Virol 155:2053–2058
Fourment M, Gibbs AJ, Gibbs MJ (2008) SWeBLAST: a sliding window web-based BLAST tool for recombinant analysis. J Virol Methods 152:98–101
Ha C, Coombs S, Revill P, Harding R, Vu M, Dale J (2008) Molecular characterization of begomoviruses and DNA satellites from Vietnam: additional evidence that the New World geminiviruses were present in the Old World prior to continental separation. J Gen Virol 89:312–326
Hollingsworth PM, Forrest LL, Spouge JL et al (2009) A DNA barcode for land plants. Proc Natl Acad Sci USA 106:12794–12797
Ilyas M, Qazi J, Mansoor S, Briddon RW (2009) Molecular characterization and infectivity of a ‘‘Legumovirus’’ (genus Begomovirus: family Geminiviridae) infecting the leguminous weed Rhynchosia minima in Pakistan. Virus Res 145:279–284
Ilyas M, Qazi J, Mansoor S, Briddon RW (2010) Genetic diversity and phylogeography of begomoviruses infecting legumes in Pakistan. J Gen Virol 91:2091–2101
Inoue-Nagata AK, Albuquerque LC, Rocha WB, Nagata T (2004) A simple method for cloning the complete begomovirus genome using the bacteriophage φ29 DNA polymerase. J Virol Methods 116:209–211
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003
Maruthi MN, Manjunatha B, Rekha AR, Govindappa MR, Colvin J, Muniyappa V (2006) Dolichos yellow mosaic virus belongs to a distinct lineage of Old World begomoviruses; its biological and molecular properties. Ann Appl Biol 149:187–195
Muhire B, Varsani A, Martin DP (2014) SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9:e108277
Navas-Castillo J, Fiallo-Olivé E, Sánchez-Campos S (2011) Emerging virus diseases transmitted by whiteflies. Annu Rev Phytopathol 49:219–248
Permingeat HR, Romagnoli MV, Vallejos RH (1998) A simple method for isolating high yield and quality DNA from cotton (Gossypium hirsutum L.) leaves. Plant Mol Biol Rep 16:1–6
Pita JS, Fondong VN, Sangare A, Otim-Nape GW, Ogwal S, Fauquet CM (2001) Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in Uganda. J Gen Virol 82:655–665
Qazi J, Ilyas M, Mansoor S, Briddon R (2007) Legume yellow mosaic viruses: genetically isolated begomoviruses. Mol Plant Pathol 8:343–348
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tiendrébéogo F, Lefeuvre P, Hoareau M, Harimalala MA, De Bruyn A, Villemot J, Traoré VSE, Konaté G, Traoré AS, Barro N, Reynaud B, Traoré O, Lett JM (2012) Evolution of African cassava mosaic virus by recombination between bipartite and monopartite begomoviruses. Virol J 9:67
Varsani A, Navas-Castillo J, Moriones E, Hernández-Zepeda C, Idris A, Brown JK, Zerbini FM (2014) Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Arch Virol 159:2193–2203
Acknowledgements
This work was supported by a grant to J. N. C. from Ministerio de Economía y Competitividad (MINECO, Spain) (AGL2013-48913-C2-1-R), co-financed by the European Regional Development Fund. E. F. O. is recipient of a “Juan de la Cierva-Incorporación” contract (MINECO, Spain). H.G.M. is recipient of a fellowship from Bill & Melinda Gates Foundation through a subcontract (B0436x12) from the Natural Resources Institute, University of Greenwich, UK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest. The research reported here did not involve experimentation with human participants or animals.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mollel, H.G., Sseruwagi, P., Ndunguru, J. et al. Desmodium mottle virus, the first legumovirus (genus Begomovirus) from East Africa. Arch Virol 162, 1799–1803 (2017). https://doi.org/10.1007/s00705-017-3289-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3289-1