
Abstract
Four isolates of a bipartite begomovirus from naturally infected Deinbollia borbonica plants exhibiting yellow mosaic symptoms in Kenya and Tanzania were molecularly characterised. The DNA-A was most closely related to that of tomato leaf curl Mayotte virus (AM701764; 82%), while the DNA-B shared the highest nucleotide sequence identity with that of East African cassava mosaic virus (AJ704953) at 65%. Based on the current ICTV species demarcation criterion for the genus Begomovirus (≥91% sequence identity for the complete DNA-A), we report the full-length genome sequence of this novel bipartite begomovirus. The results reveal additional diversity and reservoir hosts of begomoviruses in East Africa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Begomoviruses (family Geminiviridae) are an extremely successful group of emerging viruses infecting cultivated (crop), and non-cultivated (weed) plants from different botanical families [1]. The high degree of genetic variability and species diversity exhibited by begomoviruses in Africa threatens the cultivation of economically important crops such as cassava, tomato and beans [2]. Weed-infecting begomoviruses are transmitted by the polyphagous Bemisia tabaci (Gennadius) (Aleyrodidae) [3] and may act as progenitors of crop-infecting begomoviruses [4] or increase the genetic diversity of the latter by recombination [5]. Despite the critical role that weed-infecting begomoviruses play in the epidemiology of crop diseases, they remain under-studied in Africa.
Deinbollia borbonica Scheff. (Sapindaceae) is a common perennial tropical shrub whose geographical distribution spreads from the coastal belt of Somalia to northern Mozambique. In East Africa, it grows as a weed within mixed cropping farming systems where crops such as cassava, tomato, and beans are grown. Despite D. borbonica existing in Africa for over a century (https://plants.jstor.org/stable/10.5555/al.ap.specimen.m0108980), it was not until in 2012 that a begomovirus naturally infecting D. borbonica was identified in northeastern Tanzania by Ndunguru et al. (unpublished; GenBank accession no. KT799138). Here, we report the full-length genome sequence of this novel bipartite begomovirus infecting D. borbonica in Kenya and Tanzania.
Infected leaf samples showing yellow mosaic symptoms (Supplementary Fig. S1) were collected from D. borbonica in Kenya (sample K1; GPS coordinates 03.31669S, 39.96528E; altitude 12 masl; May 2014) and Tanzania (samples T1, T2, and T3; GPS coordinates 05.10219S, 38.47172E; altitude 202 masl; March 2015). Total DNA was extracted using a ZR Plant/Seed DNA MiniPrep Kit (Zymo Research Corp.). The identity of the plant species was confirmed by PCR amplification and Sanger sequencing of the chloroplast rbcL gene [6, 7]. The virus genome was enriched by rolling-circle amplification (RCA) using a TempliPhi 100 RCA Kit (GE Healthcare), and sequenced using an Illumina MiSeq system at BecA-ILRI Hub (Nairobi, Kenya). De novo assembly using CLC Genomics Workbench version 7.0.4 (CLC Bio; QIAGEN) generated two consensus contigs identified as DNA-A and DNA-B by BLASTn analysis (http://www.ncbi.nlm.nih.gov/BLAST). The genome components were amplified using abutting primers (DNA-Af, 5′-TTGGGCTCCAAGTTTTGACG-3′; DNA-Ar, 5′-TACGCGTCAAAACTTGGAGC-3′) and DNA-Bf, 5′-TGTCGTATGCGTGCTTTTGG-3′; DNA-Br, 5′-AATAGCCTCCAAAAGCACGC-3′) using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific). The blunt-ended PCR products were cloned into PUC18 and sequenced by the Sanger method using overlapping PCR primers (Supplementary Table S1).
The genome organization of the virus was identical to that of Old World bipartite begomoviruses (Supplementary Table S2). The DNA-A and DNA-B contained a 158-nucleotide (nt)-long common region (CR) with >97 % nucleotide sequence identity. Additionally, a unique putative iteron sequence (GAGGGCA), appearing twice as perfect repeats and once as an inverted repeat sequence, was identified, indicating that the cloned DNA-A and DNA-B from each plant sample constituted a cognate pair. The CR also possessed the nonanucleotide sequence (5′-TAATATTAC-3′) typical of begomoviruses [8]. Recombination analysis using RDP4 revealed no evidence of significant recombination events.
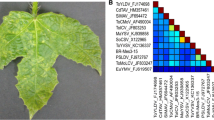
Pairwise nucleotide sequence identities calculated using Sequence Demarcation Tool (SDT v1.2) [9] revealed that the isolates were >98% identical to each other and <91% identical to currently reported begomoviruses (Supplementary Fig. S2). According to the recently updated ICTV begomovirus species demarcation criteria [10], the isolates qualify to belong to a novel begomovirus species, tentatively named “Deinbollia mosaic virus” (based on the host plant and disease symptom). Phylogenetic reconstruction based on the full-length nucleotide sequences of the DNA-A (Fig. 1A) and DNA-B (Fig. 1B) using GTR + G + I as the best-fit model of evolution in MEGA 6 software [11], clustered DMV isolates together with African begomoviruses. The DNA-A sequence was most similar (82%) to that of tomato leaf curl Mayotte virus (ToLCYTV; AM701764), while the DNA-B sequence shared 65% nucleotide sequence identity with that of East African cassava mosaic virus (EACMV: AJ704953). The results reveal additional diversity and reservoir hosts of begomoviruses in East Africa.

Maximum-likelihood (ML) trees inferred from the alignment of full-length DNA-A (A) and DNA-B (B) nucleotide sequences of DMV (in bold) and selected begomoviruses. Bootstrap values above 50% (1,000 replications) are indicated above the branches. Refer to Supplementary Table S3 for begomovirus names and acronyms
GenBank accession numbers
KT878823, KT878824, KT878825, KT878826, KT878827, KT878828, KT878829, KT878830, KT878831.
References
Seal SE, VandenBosch F, Jeger MJ (2006) Factors influencing begomovirus evolution and their increasing global significance: implications for sustainable control. CRC Crit Rev Plant Sci 25:23–46
Rey MEC, Ndunguru J, Berrie LC et al (2012) Diversity of dicotyledenous-infecting geminiviruses and their associated DNA molecules in Southern Africa, including the South-West Indian Ocean Islands. Viruses 4:1753–1791
Sseruwagi P, Maruthi MN, Colvin J et al (2006) Colonization of non-cassava plant species by cassava whiteflies (Bemisia tabaci) in Uganda. Entomol Exp Appl 119:145–153
Alabi OJ, Ogbe FO, Bandyopadhyay R et al (2008) Alternate hosts of African cassava mosaic virus and East African cassava mosaic Cameroon virus in Nigeria. Arch Virol 153:1743–1747
Nascimento LD, Silva SJC, Sobrinho RR et al (2016) Complete nucleotide sequence of a new begomovirus infecting a malvaceous weed in Brazil. Arch Virol 161:1735–1738
Levin RA, Wagner WL, Hoch PC et al (2003) Family-level relationships of Onagraceae based on chloroplast rbcL and ndhF data. Am J Bot 90:107–115
Kress WJ, Erickson DL, Jones FA et al (2009) Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proc Natl Acad Sci USA 106:18621–18626
Argüello-Astorga GR, Ruiz-Medrano R (2001) An iteron-related domain is associated to Motif 1 in the replication proteins of geminiviruses: identification of potential interacting amino acid-base pairs by a comparative approach. Arch Virol 146:1465–1485
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9:e108277
Brown JK, Zerbini FM, Navas-Castillo J et al (2015) Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch Virol 160:1593–1619
Martin DP, Murrell B, Golden M et al (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:1–5
Acknowledgements
This study was funded by the Bill & Melinda Gates Foundation (grant number OPP1052391) through Mikocheni Agricultural Research Institute (MARI), Tanzania.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Research involving human participants and/or animals
No part of this study was performed with human participants or animals by any of the authors.
Informed consent
Informed consent was obtained from individual farmers for the samples used in this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2016_3217_MOESM1_ESM.pdf
Supplementary Fig. S1 Healthy (A) and naturally infected (B) Deinbollia borbonica plants. The infected plant shows yellow mosaic symptoms and distortion of leaves (PDF 184 kb)
705_2016_3217_MOESM2_ESM.pdf
Supplementary Fig. S2 Nucleotide sequence identity plot of the full-length DNA-A of DMV and reference begomoviruses calculated using Sequence Demarcation Tool (SDT) v. 1.2 (PDF 43 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kyallo, M., Sseruwagi, P., Skilton, R.A. et al. Deinbollia mosaic virus: a novel begomovirus infecting the sapindaceous weed Deinbollia borbonica in Kenya and Tanzania. Arch Virol 162, 1393–1396 (2017). https://doi.org/10.1007/s00705-016-3217-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3217-9