
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) has caused significant economic losses in the swine industry worldwide. However, there is not an ideal vaccine to provide complete protection against PRRSV. Thus, the need for new antiviral strategies to control PRRSV still remains. Surfactant protein A (SP-A) belongs to the family of C-type lectins, which can exert antiviral activities. In this present study, we assessed the antiviral properties of recombinant porcine SP-A (RpSP-A) on PRRSV infection in Marc 145 cells and revealed its antiviral mechanism using a plaque assay, real-time qPCR, western blotting analysis and an attachment and penetration assay. Our results showed that RpSP-A could inhibit the infectivity of PRRSV in Marc 145 cells and could reduce the total RNA and protein level. The attachment assay indicated that RpSP-A in the presence of Ca2+ could largely inhibit Marc 145 cell attachment; however, in the penetration assay, it was relatively inactive. Furthermore, our study suggested that virus progeny released from infected Marc145 cells were blocked by RpSP-A from infecting other cells. We conclude that RpSP-A has antiviral activity against PRRSV, most probably by blocking viral attachment and the cell-to-cell transmission pathway, and therefore, RpSP-A holds promise as a novel antiviral agent against PRRSV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) infection is common worldwide and has become one of the most economically important diseases in the swine industry [1]. PRRSV can cause reproductive failure in sows and is associated with a respiratory disease complex that can affect pigs of all ages [2–4]. PRRSV was discovered almost simultaneously in the late 1980s on the European and North American continents and was divided into type 1 PRRSV (European genotype) and type 2 PRRSV (North American genotype) [5, 6]. PRRSV is an enveloped, single-stranded positive-sense RNA virus. Its genome is approximately 15.4 kb in size and has at least 10 open reading frames (ORFs) [7]. ORF1a and ORF1b encode non-structural proteins, and the other genes encode structural proteins, including GP2a, GP2b, GP3, GP4, GP5, GP5a, matrix protein M and the nucleocapsid protein N [8–13]. Of these, four are glycoproteins: the minor proteins GP2a, GP3 and GP4 and the major protein GP5, which is thought to be important for virus assembly and entry into permissive cells.
Researchers have made great efforts towards preventing and controlling PRRSV infection. Currently, vaccination is the main method to control PRRSV infection. Although many commercially available modified live and inactivated PRRSV vaccines have been developed, they fail to provide complete protection against the devastating disease caused by this virus [6, 14–16]. Furthermore, a large amount of literature has reported natural compounds that exhibit activity against PRRSV infection, including flavaspidic acid AB [17], matrine [18], 12-deoxyphorbol 13-phenylacetate 20-acetate (dPPA), ouabain, valinomycin and bufalin [19], and glycyrrhizin [20]. Additionally, other therapies designed to control PRRSV infection have been reported, such as LiCl [21], polyinosinic-polycytidylic acid [22], type I and type III interferons [23], double-stranded RNA (dsRNA) activated caspase oligomerizer (DRACO) [24], and microRNAs [25]. However, none of these aforementioned therapies can effectively treat or prevent PRRSV infection, so there is an urgent need to develop new antiviral strategies.
Surfactant protein A (SP-A) belongs to a family of mammalian C-type lectins, and it is an important innate immune molecule that is involved in a range of immune responses. The primary structure of SP-A is composed of four regions, including an N-terminal region, a collagen-like region, a coiled-coil neck peptide, and a carbohydrate-recognition domain (CRD) [26]. The CRD can interact with sugar moieties that are present on the surface of many pathogens, including viruses, fungi and bacteria, resulting in inhibition of infectivity in a calcium-dependent manner. Previous studies on the antiviral activity of SP-A have been focused on human medicine. In the late 1990s, human SP-A was shown to inhibit respiratory syncytial virus by blocking viral entry and subsequent syncytium formation [27]. Research on porcine SP-A activity against PRRSV was limited to one report indicating that recombinant porcine SP-A could exert anti-PRRSV activity [28]. Unfortunately, no additional studies on the anti-PRRSV potential and mechanism of SP-A were performed. However, studies on the antiviral activity of SP-D, which has a primary structure that resembles that of SP-A, have been reported more frequently. SP-D had an inhibitory effect on HIV infection through inhibition of viral entry by affecting gp120–CD4 interactions [29]. Marine and colleagues demonstrated that recombinant porcine SP-D prevented the attachment of human seasonal H1N1 and H3N2 virus to receptors on epithelial cells of the upper respiratory tract [? 36 ?]. Notably, porcine ficolin is also a collagenous lectin that was established to reduce PRRSV infection in Marc-145 cells in neutralization assays and to inhibit the replication of infectious viral particles in a GlcNAc-dependent manner [30].
In the present study, we report the antiviral properties and mechanism of action of recombinant porcine SP-A in Marc-145 cells. Our results indicate that RpSP-A affects virus attachment and blocks the cell-to-cell transmission pathway to suppress PRRSV infection in Marc-145 cells, which indicates that RpSP-A may be a useful antiviral agent for the treatment of PRRSV infection.
Materials and methods
Cells and virus
Marc 145 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10 % newborn bovine serum (NBS) at 37 °C and 5 % CO2. Spodoptera frugiperda cells (Sf9 cells), purchased from Invitrogen, were grown in Grace’s insect medium (1×; Gibco, Grand Island, NY, USA) supplemented with 10 % heat-inactivated fetal bovine serum (FBS; ScienCell, CA, USA) for recombinant baculovirus production or were grown in Sf 900 II SFM (Gibco, Grand Island, NY, USA) for protein expression at 27 °C.
HP PRRSV strain NJ-a (type 2 PRRSV) was isolated from a pig on a farm with an atypical PRRS outbreak in Nanjing city, Jiangsu province, China, in 2008. It was identified and stored by the National Research Center of Engineering and Technology for Veterinary Biologicals. The NJ-a strain was propagated and titrated by the plaque method in Marc145 cells to prepare the viral stock used in this study.
Preparation of recombinant porcine SP-A (RpSP-A)
RpSP-A was obtained using a Bac-to-Bac baculovirus expression system (Invitrogen, CA, USA). An N-terminal in-frame fusion of the pSP-A (GenBank accession no. NM_214265) gene with an 8-histidine tag was codon-optimized for baculovirus expression, synthesized, and cloned in the pUC57 vector by GenScript (Piscataway, NJ, USA).
The recombinant pSP-A gene was subcloned into a pFastBac1 vector under the control of the PH promoter. Once generated in a pFastBac™ construct, the plasmid DNA was used to transform MAX Efficiency® DH10Bac™ competent E. coli for transposition into a bacmid. Blue/white selection was used to identify colonies that contained a recombinant bacmid. High-molecular-weight RpSP-A bacmid DNA from DH10Bac™ E. coli was isolated using a BAC/PAC DNA Isolation Kit (OMEGA, USA). The concentration and purity of the RpSP-A bacmid were estimated using a Thermo Scientific NanoDropTM 2000 spectrophotometer, which was then used to transfect Sf9 insect cells using Cellfectin II reagent. The recombinant baculovirus was harvested 7 days post-transfection and was amplified twice to obtain higher titer virus stocks. Recombinant P3 viral stocks were used to infect Sf9 cells at an MOI of 1.5 PFU/cell. At 5 days postinfection, cells were harvested and washed twice in phosphate-buffered saline (PBS). Cell pellets were immediately stored at -80 °C.
The protein RpSP-A was extracted and purified using a 1-mL-size Ni2+-Sepharose HisTrap HPTM affinity column (GE Healthcare, Sweden) according to the instructions provided by the manufacturer. Imidazole in the eluate, which contained RpSP-A, was removed on a HiTrap™ desalting column (5 mL) and then exchanged with storage buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 0.1 mM PMSF). Then, the eluate was filter-sterilized through a 0.22-µm membrane filter (Millipore) and analyzed by Western blotting using an anti-His antibody (SunShineBio, China). Protein concentrations were determined using a BCA Protein Assay Reagent Kit (Pierce, Rockford, IL, USA).
Cell viability assay
The cytotoxic effect of RpSP-A was examined using an MTT assay. Briefly, Marc 145 cells in 96-well plate were exposed to various concentrations of RpSP-A at 37 °C in a 5 % CO2 atmosphere for 48 h. After incubation, the culture medium was replaced with fresh medium containing 20 µL of MTT at a concentration of 5 mg/ml, and the cells were incubated for an additional 4 h. After removal of the supernatant, 150 µL of DMSO was added to each well to dissolve the crystals for 10 min. Absorbance at 490 nm was recorded using an ELISA microplate reader.
Plaque assay
Antiviral activity was detected by plaque assay using Marc 145 cells in 6-well plates. A preliminary study was performed to determine the concentration of infectious virus particles using a 2 × 10−4 dilution of virus stock (1.4 × 106 PFU mL−1). Subconfluent monolayers of Marc145 cells in 6-well plates were treated with RpSP-A at the indicated concentrations and were simultaneously infected with PRRSV. The cells continued to be cultured at 37 °C for 1 h. Then, the cells were washed five times with PBS and covered with 2 mL of medium containing a 1:1 mixture of 2× DMEM and 2 % low-gelling-temperature agarose (Sigma, USA). After 72 h, the agarose was removed, and cells were stained with 1 % crystal violet dissolved in 10 % formaldehyde at 37 °C for 4 h. After washing gently with water, plaques were counted. Antiviral activity was calculated using the following formula: inhibition rate (%) = \( 1-\frac{\text{number of plaques}_\text{test}}{\text{number of plaques}_\text{control}} \times 100\; \% \)
Real-time quantitative PCR (real-time qPCR)
Total RNA was extracted from samples using a TaKaRa MiniBEST Universal RNA Extraction Kit (TaKaRa, Osaka, Japan). RNA was reverse transcribed into first-strand cDNA using PrimeScriptTM Reverse Transcriptase with random primers in a 20-µL final reaction volume according to the manufacturer’s instructions. First, 5 µL of total RNA was added to mixture I containing 1 µL of random primer and 4 µL of dNTPs (2.5 mM), and then the mixture was heated at 65 °C for 5 min and then kept at 4 °C for 5 min. Second, a reaction mixture was prepared by adding the above mixture to mixture II, containing 4 µL of 5× PrimeScript Buffer, 1 µL of RNase inhibitor, 1 µL of PrimeScript Reverse Transcriptase and 4 µL of RNase-free H2O. The reaction was performed under the following conditions: 30 °C for 10 min, 42 °C for 30 min, 70 °C for 15 min and, finally, 4 °C for 10 min.
Real-time qPCR was performed in a LightCycler480 II System (Roche, Basel, Switzerland) using an EvaGreen 2X qPCR MasterMix-No Dye kit (abm, Canada). Reaction mixtures were prepared according to the manufacturer’s instructions and incubated for 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C, followed by 15 s at 95 °C, 1 min at 60 °C, and 15 s at 95 °C. A portion of the ORF5 gene was detected by real-time qPCR, and GAPDH was used as a control. The data were analyzed using the 2-ΔΔCt method. Customized primers are listed in Table 1.
N protein detection by western blotting
Total protein was extracted using cell lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 % Triton X-100, 0.1 mM PMSF) and quantified using a BCA Protein Assay Reagent Kit (Pierce, Rockford, IL, USA). Then, 20 µg total protein was subjected to 12 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinyl difluoride (PVDF) membrane, which was and blocked with 5 % skim milk in TBST buffer at 37 °C for 1 h. Membranes were incubated overnight with gentle shaking at 4 °C with rabbit anti-PRRSV N protein antibody (diluted at 1:2500, donated by Dr. Yuanpeng Zhang) and mouse anti-β-actin antibody (1:200, Boster, China). After washing, membranes were incubated with an appropriate secondary antibody (1:10,000, SunShineBio, China) for 1 h at 37 °C. Antibodies were visualized using enhanced chemiluminescence (ECL) reagent (Boster, China) and images were recorded using an ImageQuant LAS 4000 (GE Healthcare, Sweden).
PRRSV attachment assay
The attachment assay was carried out as described previously [31] with some modifications. Marc 145 cells were seeded into 6-well plates. Cells were washed with cold PBS followed by DMED and then were pre-chilled at 4 °C for 30 min. Cells were incubated with PRRSV at an MOI of 0.01 and purified RpSP-A (30 µg mL−1) at 4 °C for 2 h. Cells were washed with cold PBS to remove unbound virus and RpSP-A, and then, fresh medium containing 2 % FBS was added. The cells were then cultured at 37 °C for 24 h, after which the virus titer was measured.
PRRSV penetration assay
The penetration assay was performed as described previously with some modifications [17, 31]. PRRSV could be internalized from the surface of Marc 145 cells within 3 h to 6 h [17]. Thus, we examined the kinetics of activity against PRRSV penetration using time-of-addition assays. Marc 145 cells were seeded into 6-well plates. Cells were washed with cold PBS, followed by DMED, and were then pre-chilled at 4 °C for 30 min. Cells were incubated with PRRSV at 4 °C for 2 h. Cells were washed with cold PBS. RpSP-A was added at 0 h, 1 h, and 2 h and removed at 3 h following a temperature shift. The time point when the cell culture was shifted to 37 °C was designated as 0 h. Cells were washed with cold PBS to remove unbound virus and RpSP-A, and fresh medium containing 2 % FBS was added. Cells were incubated for 24 h at 37 °C, and the virus titer was measured.
Data analysis
The data are presented as the mean ± standard deviation from three independent experiments. The data were analyzed using Student’s t-test with GraphPad Prism 6 software. The differences between the mean values were considered statistically significant when the P-value was less than 0.05.
Results
Measurement of anti-PRRSV activity of RpSP-A by plaque assay
Plaque assay results revealed that RpSP-A had potency against PRRSV. As shown in Fig. 1A, the number of visible plaques was significantly reduced in the presence of 30 µg of RpSP-A per mL. Furthermore, as shown in Fig. 1B and C, treatment with 30 µg of RpSP-A per mL reduced plaques by 77 % from 1.46 × 106 PFU mL−1 to 0.33 × 106 PFU mL−1 (P < 0.0001), whereas treatment with 15 µg of RpSP-A per mL reduced plaques by 43 % from 1.46 × 106 PFU mL−1 to 0.83 × 106 PFU mL−1 (P < 0.05). Treatment with 30 µg of RpSP-A per mL in the absence of Ca2+ was used as a negative control, and no inhibition was observed.

Anti-PRRSV activity of RpSP-A, examined using a plaque assay. A. Visible plaques on Marc 145 cells infected with PRRSV in 6-well culture plates. B. The corresponding virus titer after treatment with RpSP-A. C. Inhibition of infection by RpSP-A. Each value represents the mean of three independent experiments with standard derivation. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to the 0/Ca+ group
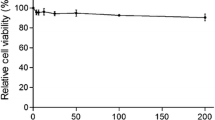
To rule out the possibility that nonspecific toxicity caused by RpSP-A was affecting PRRSV replication, we investigated its cytotoxicity in Marc-145 cells at concentrations of 0, 10, 20, 30, 40, 50, 60 µg/mL. As shown in Fig. 2, 48 hours after treatment, the cells cultured in medium containing RpSP-A at concentrations below 60 µg mL−1 retained approximately 100 % viability relative to the control.

Cytotoxicity of RpSP-A in Marc 145 cells, measured using an MTT assay. Cells were incubated with different concentrations of RpSP-A for 48 h, and a cell viability assay was performed
Effects of RpSP-A on virus attachment and penetration
To elucidate the mechanism of the antiviral effects of RpSP-A, a virus entry assay was utilized. The data showed that RpSP-A could largely inhibit virus attachment; however, it was relatively inactive against virus penetration.
In the virus attachment assay, Marc 145 cells were incubated with PRRSV at an MOI of 0.01 in the presence of RpSP-A at 4 °C, a condition in which viruses attach to cells but do not penetrate. As shown in Fig. 3, treatment with 30 µg of RpSP-A per mL largely inhibited Marc145 cell attachment. The mean viral titer was 105.64 PFU mL−1 in the absence of RpSP-A, but it was reduced to 104.03 PFU mL−1 in the presence of 30 µg of RpSP-A per mL with the addition of Ca2+ (P < 0.0001). No significant difference was observed between 0 and 30 µg mL−1 of RpSP-A in the absence of Ca2+ (P > 0.05).

Effect of RpSP-A on attachment of PRRSV to Marc 145 cells. Cells were incubated for 24 h after treatment with PRRSV alone or together with RpSP-A in the presence or absence of calcium ions, and then virus titer was measured. Data are presented as mean ± S.D. of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to the control
In the penetration assay, we studied the inhibition kinetics of RpSP-A against PRRSV. The mean viral titer in the presence of 30 µg of RpSP-A per mL with the addition of Ca2+ was 104.56 PFU mL−1 at 0 h (P < 0.0001), 105.3 PFU mL−1 at 1 h (P < 0.05), and 105.46 PFU mL−1 at 2 h (P > 0.05) compared to 105.6 PFU mL−1 in the absence of RpSP-A (Fig. 4), suggesting that the inhibitory effect of RpSP-A on virus penetration decreased over time, although there was no significant difference at 2 h. Additionally, 30 µg of RpSP-A per mL in the absence of Ca2+ did not affect virus penetration.

Effect of RpSP-A on Marc 145 cell penetration. Cells were incubated for 24 h after treatment with PRRSV alone or after addition of RpSP-A at 1-h intervals in the presence or absence of calcium ions, and then virus titer was measured. Data are presented as mean ± S.D. of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to the control
Results of RT-PCR and western blotting
We next attempted to investigate the kinetics of the RpSP-A-mediated effect on the intracellular viral load. First, Marc 145 cells were incubated with PRRSV at an MOI of 0.01 for 24 h, and the medium was replaced with fresh medium containing 30 µg of RpSP-A per mL. Then, the total RNA in Marc 145 cells was extracted for PRRSV RNA genome detection by real-time RT-PCR after culture for 6 h, 12 h, 24 h or 36 h. As shown in Fig. 5A, the addition of RpSP-A resulted in a significant reduction in the total RNA level at 12 h, 24 h and 36 h compared with the non-drug-treated cells (P < 0.0001), and this reduction reached a peak at 24 h after addition of RpSP-A. However, there was no significant difference at 6 h following RpSP-A addition compared with the non-drug-treated group (P > 0.05). Moreover, we also detected N protein of PRRSV in Marc 145 cells by western blotting after treatment with RpSP-A for 36 h. As shown in Fig. 5B, western blotting analysis indicated that RpSP-A reduced the level of N protein in cells in a dose-dependent manner.

Reduction of the total levels of PRRSV RNA and protein of in Marc 145 cells by treatment with RpSP-A. A. The relative mRNA level was determined by amplifying a portion of PRRSV ORF5 using real-time RT-PCR at different time points. Relative expression (fold change) in comparison to the control group in the absence of RpSP-A (set as 1) is illustrated. Data are presented as mean ± S.D. of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to the control. B. The viral N protein and β-actin were detected by western blotting. β-actin was used as an internal control
Discussion
Surfactant protein A, an antimicrobial defense molecule of the innate immune system, has the potential to be exploited as an antiviral drug in the future. For this reason, we sought to develop it as a recombinant product in this study. Here, RpSP-A was obtained using a Bac-to-Bac baculovirus expression system. We found that RpSP-A expressed by Sf9 insect cells could block virus entry. Viral entry represents an attractive target for chemotherapeutic intervention [32]. However, drugs that inhibit virus entry prevent infection of cells by cell-free virus particles and also prevent virus transmission between virus-infected and uninfected cells [32, 33]. The capacity to inhibit both modes of infection by a single drug is one of the reasons why SP-A may be superior to other microbicide drugs. Thus, SP-A might be a candidate agent for preventing PRRSV infection.
So far, there have been limited data available regarding the anti-PRRSV activity of porcine SP-A. In China, highly pathogenic PRRSV appears to be widely prevalent, and therefore, the highly pathogenic PRRSV strain NJ-a, a type 2 PRRSV strain, was used in this study. To explore the anti-PRRSV activity of porcine SP-A, further studies need to be performed on type 1 PRRSV. Our data indicate that RpSP-A significantly suppresses PRRSV infection in a dose-dependent manner without causing cytotoxicity in Marc 145 cells. We also found that RpSP-A exerts potent antiviral activity in a Ca2+-dependent manner, indicating that RpSP-A may interact with PRRSV via the carbohydrate-recognition domain, which is consistent with previous studies [27, 28].
Researchers have confirmed that lectins inhibit virus entry [27, 29, 34, 35], and each step of the viral entry pathway is a potential target for antiviral agents. We conducted virus attachment and penetration assays to assess whether RpSP-A also interfered with PRRSV entry. The virus entry assay showed that RpSP-A strongly inhibits virus attachment, but its effect on virus penetration was, at most, modest, suggesting that RpSP-A most probably blocks virus attachment, like other lectins. It has been shown that hippeastrum hybrid agglutinin (HHA), a mannose-specific lectin, probably interferes with SARS coronavirus attachment. When attachment occurred at 4 °C, HHA was twice as active (EC50 = 2.5 µg mL−1) as it was in a situation where both attachment and penetration were allowed to occur at 37 °C (EC50 = 5.2 µg mL−1) [34]. In another study, recombinant porcine SP-D was shown to inhibit IAV attachment using human trachea tissue, and it fully prevented binding of human seasonal H1N1 and H3N2 IA at higher dose [35]. Our data indicate that the suppression of PRRSV penetration by RpSP-A gradually weakened and even disappeared as the time between infection and addition of RpSP-A increased, suggesting that RpSP-A might act on PRRSV entry into Marc 145 cells only at the cell surface and that it has no effect once the virus has entered the cell.
A better understanding of the underlying mechanisms by which RpSP-A influences viral entry might lead to new antiviral interventions. Taking the previously reported data and our own findings together, we hypothesize that RpSP-A inhibits PRRSV entry by binding to glycoprotein GP2, GP3, GP4 or GP5 on the surface of PRRSV, thereby preventing it from interacting with its receptors on Marc 145 cells. The mechanism of the antiviral activity of lectins has been studied by several researchers. It has been shown that human SP-A can bind to the F (fusion) glycoprotein on the surface of respiratory syncytial virus and may neutralize RSV by directly inhibiting the fusion function of this protein, thereby inhibiting viral entry [27]. In another study, Marine et al. reported that the interaction between RpSP-D and the viral HA prevented attachment of the virus to target cells and contributed to the neutralizing effects of RpSP-D [35]. In addition, other lectins such as SP-D and plant lectins have been shown to bind to glycoprotein gp120 of HIV-1 and block access of gp120 to the receptor protein CD4 [29, 32, 33, 36], thereby blocking viral entry to reduce HIV-1 infection. The nature of the interactions among PRRSV, RpSP-A and cells is not yet known, so this model should be assessed in further studies.
Interestingly, our data also indicated that RpSP-A had no effect on the total RNA level when the virus was allowed to undergo only a single cycle of replication but caused the total RNA level to decrease greatly at 12 h, 24 h and 36 h. A possible explanation for this is that virus progeny released from infected Marc145 cells are blocked by RpSP-A and prevented from infecting other cells.
Since Marc-145 cells are not of porcine origin but of monkey origin, the mechanism of infection of porcine alveolar macrophages (PAMs) is not identical to that in Marc 145 cells [37, 38]; furthermore, a previous study showed that some lectins had a stronger inhibitory effect on virus infection when PAMs were treated with lectins than when the virus was treated with lectins prior to infection, in contrast to what was observed with Marc 145 cells, suggesting potentially complex interactions among PAMs, lectins and PRRSV [39]. More studies will be needed to characterize the antiviral activity of RpSP-A in PAMs, which are the main target cells for PRRSV in vivo.
In conclusion, we show that RpSP-A exerts potent activity against HP-PRRSV in Marc 145 cells. We clearly demonstrate that RpSP-A interacts at the level of virus attachment and blocks the cell-to-cell transmission pathway, which has never been observed before. RpSP-A most probably interacts with the glycoproteins of PRRSV, thereby preventing it from binding to host-cell receptors and subsequently interfering with transmission. A better understanding of the mechanisms by which RpSP-A influences viral infection in porcine alveolar macrophages might lead to new antiviral interventions.
References
Lyoo YS (2015) Porcine reproductive and respiratory syndrome virus vaccine does not fit in classical vaccinology. Clin Exp Vaccine Res 4(2):159–165
Rossow KD (1998) Porcine reproductive and respiratory syndrome. Vet Pathol 35(1):1–20
Costers S, Delputte PL, Nauwynck HJ (2006) Porcine reproductive and respiratory syndrome virus-infected alveolar macrophages contain no detectable levels of viral proteins in their plasma membrane and are protected against antibody-dependent, complement-mediated cell lysis. J Gen Virol 87(Pt 8):2341–2351
Wei C, Huang Z, Sun L, Xie J, Chen Y, Zhang M, Zhang C, Qi H, Qi W, Ning Z, Yuan L, Wang H, Zhang L, Zhang G (2013) Expression and antibody preparation of GP5a gene of porcine reproductive and respiratory syndrome virus. Indian J Microbiol 53(3):370–375
Allende R, Lewis TL, Lu Z, Rock DL, Kutish GF, Ali A, Doster AR, Osorio FA (1999) North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J Gen Virol 80(Pt 2):307–315
Renukaradhya GJ, Meng XJ, Calvert JG, Roof M, Lager KM (2015) Inactivated and subunit vaccines against porcine reproductive and respiratory syndrome: current status and future direction. Vaccine 33(27):3065–3072
Chand RJ, Trible BR, Rowland RR (2012) Pathogenesis of porcine reproductive and respiratory syndrome virus. Curr Opin Virol 2(3):256–263
Meulenberg JJ, Petersen-den Besten A, De Kluyver EP, Moormann RJ, Schaaper WM, Wensvoort G (1995) Characterization of proteins encoded by ORFs 2 to 7 of Lelystad virus. Virology 206(1):155–163
Firth AE, Zevenhoven-Dobbe JC, Wills NM, Go YY, Balasuriya UB, Atkins JF, Snijder EJ, Posthuma CC (2011) Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J Gen Virol 92(Pt 5):1097–1106
Snijder EJ, van Tol H, Pedersen KW, Raamsman MJ, de Vries AA (1999) Identification of a novel structural protein of arteriviruses. J Virol 73(8):6335–6345
Meulenberg JJ, Petersen-den Besten A (1996) Identification and characterization of a sixth structural protein of Lelystad virus: the glycoprotein GP2 encoded by ORF2 is incorporated in virus particles. Virology 225(1):44–51
Wu WH, Fang Y, Farwell R, Steffen-Bien M, Rowland RR, Christopher-Hennings J, Nelson EA (2001) A 10-kDa structural protein of porcine reproductive and respiratory syndrome virus encoded by ORF2b. Virology 287(1):183–191
Johnson CR, Griggs TF, Gnanandarajah J, Murtaugh MP (2011) Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J Gen Virol 92(Pt 5):1107–1116
Renukaradhya GJ, Meng XJ, Calvert JG, Roof M, Lager KM (2015) Live porcine reproductive and respiratory syndrome virus vaccines: current status and future direction. Vaccine 33(33):4069–4080
Scortti M, Prieto C, Alvarez E, Simarro I, Castro JM (2007) Failure of an inactivated vaccine against porcine reproductive and respiratory syndrome to protect gilts against a heterologous challenge with PRRSV. Vet Rec 161(24):809–813
Zuckermann FA, Garcia EA, Luque ID, Christopher-Hennings J, Doster A, Brito M, Osorio F (2007) Assessment of the efficacy of commercial porcine reproductive and respiratory syndrome virus (PRRSV) vaccines based on measurement of serologic response, frequency of gamma-IFN-producing cells and virological parameters of protection upon challenge. Vet Microbiol 123(1–3):69–85
Yang Q, Gao L, Si J, Sun Y, Liu J, Cao L, Feng W-H (2013) Inhibition of porcine reproductive and respiratory syndrome virus replication by flavaspidic acid AB. Antiviral Res 97(1):66–73
Sun N, Wang Z-W, Wu C-H, Li E, He J-P, Wang S-Y, Hu Y-L, Lei H-M, Li H-Q (2014) Antiviral activity and underlying molecular mechanisms of Matrine against porcine reproductive and respiratory syndrome virus in vitro. Res Vet Sci 96(2):323–327
Karuppannan AK, Wu KX, Qiang J, Chu JJ, Kwang J (2012) Natural compounds inhibiting the replication of Porcine reproductive and respiratory syndrome virus. Antiviral Res 94(2):188–194
Duan E, Wang D, Fang L, Ma J, Luo J, Chen H, Li K, Xiao S (2015) Suppression of porcine reproductive and respiratory syndrome virus proliferation by glycyrrhizin. Antiviral Res 120:122–125
Hao HP, Wen LB, Li JR, Wang Y, Ni B, Wang R, Wang X, Sun MX, Fan HJ, Mao X (2015) LiCl inhibits PRRSV infection by enhancing Wnt/beta-catenin pathway and suppressing inflammatory responses. Antiviral Res 117:99–109
Zhang L, Liu J, Bai J, Du Y, Wang X, Liu X, Jiang P (2013) Poly(I:C) inhibits porcine reproductive and respiratory syndrome virus replication in MARC-145 cells via activation of IFIT3. Antiviral Res 99(3):197–206
Luo R, Fang L, Jin H, Jiang Y, Wang D, Chen H, Xiao S (2011) Antiviral activity of type I and type III interferons against porcine reproductive and respiratory syndrome virus (PRRSV). Antiviral Res 91(2):99–101
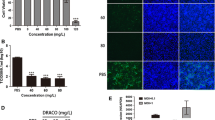
Guo C, Chen L, Mo D, Chen Y, Liu X (2015) DRACO inhibits porcine reproductive and respiratory syndrome virus replication in vitro. Arch Virol 160(5):1239–1247
Zhang Q, Guo XK, Gao L, Huang C, Li N, Jia X, Liu W, Feng WH (2014) MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology 450–451:182–195
Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, Bernal AL, Reid KB, Madan T, Chakraborty T (2006) Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol 43(9):1293–1315
Ghildyal R, Hartley C, Varrasso A, Meanger J, Voelker DR, Anders EM, Mills J (1999) Surfactant protein A binds to the fusion glycoprotein of respiratory syncytial virus and neutralizes virion infectivity. J Infect Dis 180(6):2009–2013
Zhang F, Zhong F, Li X, Wang X, Zhang K, Chen H, Li Z, Li W, Pan H, Han D (2012) Recombinant porcine lung surfactant protein A inhibits porcine reproductive and respiratory syndrome virus infection into host cells in vitro. Wei Sheng Wu Xue Bao 52(5):654–660
Pandit H, Gopal S, Sonawani A, Yadav AK, Qaseem AS, Warke H, Patil A, Gajbhiye R, Kulkarni V, Al-Mozaini MA, Idicula-Thomas S, Kishore U, Madan T (2014) Surfactant protein D inhibits HIV-1 infection of target cells via interference with gp120-CD4 interaction and modulates pro-inflammatory cytokine production. PloS one 9(7):e102395
Keirstead ND, Lee C, Yoo D, Brooks AS, Hayes MA (2008) Porcine plasma ficolin binds and reduces infectivity of porcine reproductive and respiratory syndrome virus (PRRSV) in vitro. Antiviral Res 77(1):28–38
Xiang Y, Pei Y, Qu C, Lai Z, Ren Z, Yang K, Xiong S, Zhang Y, Yang C, Wang D, Liu Q, Kitazato K, Wang Y (2011) In vitro anti-herpes simplex virus activity of 1,2,4,6-tetra-O-galloyl-β-d-glucose from Phyllanthus emblica L. (Euphorbiaceae). Phytother Res 25:975–982
Balzarini J (2006) Large-molecular-weight carbohydrate-binding agents as HIV entry inhibitors targeting glycoprotein gp120. Curr Opin HIV AIDS 1(5):355–360
Balzarini J (2006) Inhibition of HIV entry by carbohydrate-binding proteins. Antiviral Res 71(2–3):237–247
Keyaerts E, Vijgen L, Pannecouque C, Van Damme E, Peumans W, Egberink H, Balzarini J, Van Ranst M (2007) Plant lectins are potent inhibitors of coronaviruses by interfering with two targets in the viral replication cycle. Antiviral Res 75(3):179–187
Hillaire MLB, van Eijk M, van Trierum SE, van Riel D, Saelens X, Romijn RA, Hemrika W, Fouchier RAM, Kuiken T, Osterhaus ADME, Haagsman HP, Rimmelzwaan GF (2011) Assessment of the antiviral properties of recombinant porcine SP-D against various influenza A viruses in vitro. PloS One 6(9)
Bertaux C, Daelemans D, Meertens L, Cormier EG, Reinus JF, Peumans WJ, Van Damme EJ, Igarashi Y, Oki T, Schols D, Dragic T, Balzarini J (2007) Entry of hepatitis C virus and human immunodeficiency virus is selectively inhibited by carbohydrate-binding agents but not by polyanions. Virology 366(1):40–50
Duan X, Nauwynck HJ, Pensaert MB (1997) Effects of origin and state of differentiation and activation of monocytes/macrophages on their susceptibility to porcine reproductive and respiratory syndrome virus (PRRSV). Arch Virol 142:2483–2497
Nauwynck HJ, Duan X, Favoreel HW, Van Oostveldt P, Pensaert MB (1999) Entry of porcine reproductive and respiratory syndrome virus into porcine alveolar macrophages via receptor-mediated endocytosis. J Gen Virol 80(Pt 2):297–305
Li J, Murtaugh MP (2015) Functional analysis of porcine reproductive and respiratory syndrome virus N-glycans in infection of permissive cells. Virology 477:82–88
Baughman RP, Sternberg RI, Hull W, Buchsbaum JA, Whitsett J (1993) Decreased surfactant protein A in patients with bacterial pneumonia. Am Rev Respir Dis 147(3):653–657
Acknowledgments
This study was supported by grants from the Special Fund for Agro-Scientific Research in the Public Interest (No. 201303046) and the Independent Innovation of Agricultural Sciences Program of Jiangsu Province (No. cx (14)2089).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was supported by grants from the Special Fund for Agro-Scientific Research in the Public Interest (No. 201303046) and the Independent Innovation of Agricultural Sciences Program of Jiangsu Province (No. cx (14)2089).
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Li, L., Zheng, Q., Zhang, Y. et al. Antiviral activity of recombinant porcine surfactant protein A against porcine reproductive and respiratory syndrome virus in vitro . Arch Virol 161, 1883–1890 (2016). https://doi.org/10.1007/s00705-016-2838-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2838-3