
Abstract
Background
It is suspected that microbiome-derived trimethylamine N-oxide (TMAO) may enhance platelet responsiveness and accordingly be thrombophilic. The purpose of this prospective observational study is to evaluate TMAO in patients with subarachnoid hemorrhage (SAH) and compare it with a control group. A secondary aim was to investigate TMAO in the cerebrospinal fluid (CSF) from SAH patients. This should provide a better understanding of the role of TMAO in the pathogenesis of SAH and its thrombotic complications.
Methods
The study included patients with diagnosed spontaneous SAH recruited after initial treatment on admission and patients with nerve, nerve root, or plexus disorders serving as controls. Blood samples were gathered from all patients at recruitment. Additionally, sampling of SAH patients in the intensive care unit continued daily for 14 days. The CSF was collected out of existing external ventricular drains whenever possible.
Results
Thirty-four patients diagnosed with SAH, and 108 control patients participated in this study. Plasma TMAO levels at baseline were significantly lower in the SAH group (1.7 μmol/L) compared to the control group (2.9 μmol/L). TMAO was detectable in the CSF (0.4 μmol/L) and significantly lower than in plasma samples of the SAH group at baseline. Plasma and CSF TMAO levels correlated positively. The TMAO levels did not differ significantly during the observation period of 15 days.
Conclusions
Although we assumed that patients with higher TMAO levels were at higher risk for SAH a priori, plasma TMAO levels were lower in patients with SAH compared with control subjects with nerve, nerve root, or plexus disorders on admission to the hospital. A characteristic pattern of plasma TMAO levels in patients with SAH was not found.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spontaneous subarachnoid hemorrhage (SAH) remains a major issue in patient care. It accounts for about 5% of all strokes with an incidence of around 6–9 per 100,000 per year worldwide [10, 12]. Younger people, with a mean age of around 55 years, are often affected by SAH [28]. A meta-analysis shows mortality of up to 66.7%, while up to 33% of the surviving patients are no longer able to return to work [26, 27]. Besides the initial bleeding, especially complications during the further course affect the outcome despite substantial progress in neurointensive care [16]. Delayed cerebral ischemia (DCI), for example, describes ischemia emerging 4 to 14 days after bleeding. Yet, the genesis of DCI is not well understood. In addition to vasospasm of the cerebral arteries, vascular injury caused by platelet aggregation and enhancing microvascular thrombus formation are discussed as possible mechanisms of DCI [44]. Such microvascular thrombi are found in patients with SAH and are presumed to be a part of DCI genesis. To date, no effective therapy has been elucidated against microvascular thrombus formation in SAH patients [5]. Hence, prediction and prevention are of utmost importance for both the management of SAH itself and its complications. The applicability of potential biomarkers such as trimethylamine N-oxide (TMAO) could contribute to this. The latter is known to be increased in cardiovascular risk patients and may be either a biomarker or even a mediator [17]. It is suspected that TMAO may enhance platelet responsiveness and accordingly be thrombophilic [53]. It has been associated with various diseases such as heart failure and stroke [35, 42, 43]. Contrary changes in TMAO levels have been reported in stroke patients, and the influence of TMAO on stroke patients has not yet been clarified. A separate observation of each form of a stroke may be reasonable. To the best of our knowledge, a possible impact of TMAO on SAH, in particular, has not been sufficiently explored. This study aimed to investigate the TMAO levels of patients in the acute phase of SAH and the course over the following days in the intensive care unit. The blood-CSF barrier (BCB) disturbance occurring in SAH prompted us to also investigate the presence of TMAO in cerebrospinal fluid (CSF) [25].
Materials and methods
Eligibility and study design
A prospective observational study including patients treated at the Department of Neurosurgery, University of Leipzig Medical Center (Leipzig, Germany), was performed from October 2018 to January 2020. The two study arms consisted of patients with diagnosed SAH treated at the intensive care unit (SAH group) and patients with nerve, nerve root, or plexus disorders treated at the normal unit serving as controls.
Patients with suspected non-traumatic SAH were screened for the SAH group. Patients with traumatic SAH or an age < 18 years were excluded from this study group. Patients with aneurysmal SAH and perimesencephalic SAH were considered as non-traumatic SAH. The SAH was confirmed by a neurosurgeon or neuroradiologist via cranial computed tomography followed by an intra-arterial selective digital subtraction angiography. Patients were treated either interventional with coiling or operative clipping of any aneurysms detected or conservatively with basic neuroprotective measures. Hydrocephalus was treated by external ventricular drainage if necessary, and all patients were monitored in the intensive care unit to control intracranial pressure (< 15 mmHg) and maintain sufficient cerebral perfusion pressure (> 70 mmHg). Patients do not routinely receive fibrinolytic substances, such as tranexamic acid, or the like. Nimodipine for the prophylaxis of delayed cerebral ischemia was started after initial management and at the earliest on day 1 after SAH. Transcranial Doppler examinations were performed daily to monitor blood flow velocities in the basal cerebral arteries. If a critical increase in blood flow velocities was observed, cranial computed tomography/computed tomography perfusion was performed to confirm a decrease in cerebral blood flow, and digital subtraction angiography was indicated. In the case of cerebral vasospasm being detected, inter alia, intraarterial treatment with nimodipine was started.
Patients with nerve, nerve root, or plexus disorders were screened for the control group. Patients with a history of proliferative processes, known damage to the central nervous system or an age < 18 years were excluded from this study group. The patient group that served as controls was selected from the patient population of the Department of Neurosurgery, University of Leipzig Medical Center (Leipzig, Germany), based on availability. We found no evidence for altered TMAO levels in this patient group [14, 40]. In addition, we did not expect any thrombotic events or cerebral abnormalities before treatment that could have affected the objective of the study.
Study aims
The primary aim was to observe the difference in plasma TMAO levels between the SAH group at baseline and the control group.
Secondary aims were to observe the relationship between plasma TMAO and CSF TMAO, the association between plasma TMAO and the clinical severity of SAH, and the course of TMAO after SAH, and to identify possible factors influencing plasma TMAO levels.
Sampling and data collection
Sampling for the SAH group started on the day of admission for suspected SAH (hereinafter referred to as “day 0 after SAH”) after the initial treatment. Whole blood samples were collected daily over 15 days into tubes containing ethylenediaminetetraacetic acid. Sampling stopped after 15 days, with the patient’s death or release from the intensive care unit. CSF was collected out of existing external ventricular drains where possible on admission and days 5 and 10 after SAH. Whole blood samples were collected from the control group only on admission into tubes containing ethylenediaminetetraacetic acid.
Additionally, hematocrit, hemoglobin, red blood cell count, platelet count, estimated glomerular filtration rate (eGFR), prothrombin time, and activated partial thromboplastin time were routinely measured for both patient groups. Therefore, samples were collected into tubes containing ethylenediaminetetraacetic acid, polyacrylate, or citrate, respectively.
Medical information was gathered anamnestically from the patients or their relatives or from existing medical records. The following patient monitoring data were gathered from the intensive care unit: the form of nutrition (sober, parenteral, enteral, oral), sedation status (sedated, not sedated), and intubation status (intubated, not intubated). The SAH was classified in all patients at baseline by a neurosurgical specialist according to the World Federation of Neurosurgical Societies (WFNS) classification for SAH [34]. Cranial computed tomography images obtained on admission were used by a neurosurgical specialist to grade patients according to the Fisher grading for SAH [13].
Albumin was measured in the CSF and plasma samples to calculate the CSF/plasma albumin ratio (QALB). The appearance of BCB disturbances was defined by an age-dependent upper limit of the QALB < (4 + age /15) [33].
Laboratory procedures
Measurements were performed at the Institute of Laboratory Medicine, Clinical Chemistry and Molecular Diagnostics, University of Leipzig Medical Center (Leipzig, Germany). Briefly, TMAO, betaine, carnitine, and choline plasma levels were determined by high-performance liquid chromatography-tandem mass spectrometry (LC–MS/MS). Samples were prepared by protein precipitation adding 90 μl of acetonitrile, including the internal standards, to 10 μl of the sample. After thorough mixing and centrifugation, 10 μl of the supernatant was diluted with 990 μl of eluent B. An amount of 5 μl of this mixture was used for liquid chromatography-tandem mass spectrometry analysis (a detailed method description can be found in Supplement, Expanded materials and methods). The method described for quantification of TMAO and its precursors has been used in previous studies [36], and recently a publication with the exact method description and validation has been published [8].
Hematocrit, hemoglobin, red blood cell count, and platelet count were measured on a Sysmex XN9000 analyzer (Sysmex Corporation, Kobe, Japan), according to the manufacturer’s instructions. The eGFR was estimated by measuring the serum creatinine enzymatically on the Cobas 8000 platform (Roche, Basel, Switzerland) using the Creatinine Plus assay (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Prothrombin time was measured on the ACL top 700 platform (Instrumentation Laboratory, Bedford, USA) using the RecombiPlasTin 2G assay (Instrumentation Laboratory, Bedford, USA), according to the manufacturer’s instructions. The activated partial thromboplastin time was measured on the ACL top 700 platform (Instrumentation Laboratory, Bedford, USA) using the synthASil assay (Instrumentation Laboratory, Bedford, USA), according to the manufacturer’s instructions. Albumin was analyzed in plasma and CSF using a commercially available test kit on a Roche Cobas C 111 Clinical Chemistry analyzer (Roche Diagnostics, Mannheim, Germany).
Statistical analysis
Continuous variables are presented as medians (interquartile range), unless specified otherwise, whereas categorical and dichotomous variables are presented as numbers and percentages. Variables were tested for normal distribution using the Shapiro–Wilk test. Differences between sampling times were calculated using the Wilcoxon test for non-normally distributed samples, whereas differences between the SAH group and the control group were calculated using the Mann–Whitney U test for non-normally distributed continuous variables and the Chi-square test for dichotomous variables. p-values of less than 0.05 were considered statistically significant. Correlations between parameters were calculated using Spearman’s rank test for non-normally distributed data. Statistical calculations were performed using IBM SPSS statistic 20 (Armonk, NY, USA). For post hoc power analysis, GPower 3.1.9.7 (Dusseldorf, Germany) was used.
Results
Baseline characteristics
We screened 54 patients with non-traumatic SAH and 135 patients with diagnosed nerve, nerve root, or plexus disorders. A total of 34 patients with diagnosed SAH or their legal representatives and 108 patients with nerve, nerve root, or plexus disorders agreed to recruitment.
Twenty-seven patients with SAH were treated interventionally and 7 patients conservatively. We gathered plasma samples from all patients on admission. External ventricular drains were laid in 17 patients with SAH, and CSF was collected on day 0 after SAH. Nine patients developed DCI during the observation period.
Characteristics at baseline were balanced between the two groups regarding sex, age, BMI, smoking status, and the prevalence of diabetes mellitus (Table 1). Hypertension occurred more often in the SAH group (79.4%) compared to the control group (52.8%). All SAH patients stated that they ate an omnivore diet.
Hematocrit, hemoglobin, and red blood cell count on admission were significantly lower in the SAH group compared to the control group. Prothrombin time and activated partial thromboplastin time were significantly lower in the SAH group but within the reference range. The eGFR was higher in the SAH group but was above the reference threshold for both groups.
TMAO and precursors at baseline
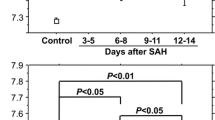
Plasma TMAO levels were significantly lower in the SAH group at baseline compared to the control group (1.7 versus 2.9 μmol/L; Fig. 1). Post hoc power analysis for the Mann–Whitney U test yielded a power of 0.90 with an effect size of d = 0.659, alpha = 0.05, and the corresponding number of cases.

Differences in TMAO levels between SAH and control group at baseline. Values are presented as median with a 95% confidence interval. p-values were calculated using Mann–Whitney U test for non-normally distributed continuous variables. The SAH indicates subarachnoid hemorrhage and TMAO, trimethylamine N-oxide
The CSF TMAO levels in the SAH group were fourfold lower than plasma TMAO levels at baseline (0.4 versus 1.7 μmol/L). Fifteen patients with SAH had a BCB disturbance defined by the QALB. The QALB did not correlate significantly with CSF TMAO levels or the CSF/plasma TMAO ratio (QTMAO). The subgroups divided by the appearance of a BCB disturbance did not differ significantly. Moreover, plasma TMAO levels correlated positively with the corresponding CSF TMAO levels (p < 0.001) (Fig. 2), whereas correlations between the CSF TMAO and its precursors in plasma samples were less high or not significant (Supplemental Fig. 1).

TMAO concentration in plasma and corresponding CSF sample in the SAH group at baseline. The SAH indicates a subarachnoid hemorrhage; CSF, cerebrospinal fluid; and TMAO, trimethylamine N-oxide
The TMAO precursors betaine, carnitine, and choline showed similar effects. Plasma levels were significantly lower in SAH patients compared to control patients. The CSF levels were significantly lower than plasma levels in the SAH group (betaine: 3.4 versus 20.3 μmol/L; carnitine: 1.4 versus 34.9 μmol/L; choline: 2.4 versus 4.5 μmol/L). Furthermore, CSF levels also correlated positively with corresponding plasma levels for all three precursors (Supplemental Fig. 1).
Patients who developed DCI had only significantly lower plasma carnitine levels than patients who did not develop DCI. Plasma and CSF TMAO levels did not differ between the DCI and non-DCI subgroups (Supplemental Table 1; Supplemental Fig. 2).
When evaluating subgroups defined by the WFNS classification, plasma TMAO levels did not differ significantly between these groups (Supplemental Table 2). Regrouping these subgroups into a good grade (WFNS classification 1–3) and a poor grade (WFNS classification 4–5), plasma TMAO levels tended to be lower in the poor grade subgroup, although the difference was not significant (Supplemental Table 3). No significant differences in plasma TMAO levels between the subgroups were found after dividing the SAH group by Fisher grading into 4 subgroups (Supplemental Table 4).
When comparing male and female patients across the groups of SAH and controls combined, males show significantly higher TMAO (2.8 versus 2.6 μmol/L), betaine (30.9 versus 21.7 μmol/L), carnitine (44.7 versus 37.9 μmol/L), and choline (6.6 versus 5.3 μmol/L) concentrations in plasma at baseline.
Dynamic of TMAO after SAH
Isolated failures in sampling occurred due to pronounced intensive care measures. The sample count decreased over the observation process because of discharge or death (Supplemental Table 5). A total of 390 blood samples were taken from all patients with SAH at all sampling times combined. Plasma TMAO levels did not vary significantly over 15 days after SAH (Fig. 3), and we did not observe a characteristic course. The eGFR varies significantly over the 15 days but was never below the reference threshold (> 90 mL/min/1.73 m2). The platelet count increased significantly after around 4 days and increased continuously until the end of the observation period. Plasma betaine levels increased in the days after SAH and become significantly higher compared to day 0 on the 7th day after SAH, while plasma carnitine and choline levels increased on the 5th day (Fig. 3, Supplemental Fig. 3). Since the absolute values of TMAO and its precursors were sometimes highly scattered, we observed the percentage changes of TMAO and its precursors, defining the value on day 0 after SAH as 100%. We did not find any considerably different courses compared to the absolute plasma levels (Supplemental Fig. 4). A total of 40 CSF samples were taken from 17 patients from the SAH group at all sampling times combined. The CSF TMAO levels did not vary significantly between days 0, 5, and 10 after SAH. Betaine, carnitine, and choline CSF levels increased significantly after day 0 (Fig. 3, Supplemental Fig. 3).

Changes in the median concentration of TMAO and choline in plasma and CSF samples and of eGFR and the platelet count in plasma samples of the SAH group over the observation period. Values are presented as median with a 95% confidence interval at each sampling time. p-values of continuous variables were calculated using the Wilcoxon test. *p ≤ 0.05 compared to the previous day; **p ≤ 0.01 compared to the previous day; ***p ≤ 0.001 compared to the previous day. ▲p ≤ 0.05 compared to day 0 after SAH; ▲▲p ≤ 0.01 compared to day 0 after SAH; and ▲▲▲p ≤ 0.001 compared to day 0 after SAH. The CSF indicates cerebrospinal fluid; eGFR, estimated glomerular filtration rate; SAH, subarachnoid hemorrhage; TMAO, trimethylamine N-oxide
Plasma TMAO levels correlated significantly with hemoglobin, platelet count, eGFR, and activated partial thromboplastin time over all 15 visit times (Fig. 4). No significant differences in the plasma TMAO, betaine, carnitine, or choline levels between associated subgroups could be detected when comparing subgroups defined by the form of nutrition, sedation status, and intubation status on each day separately (Supplemental Fig. 5).

Correlation between TMAO, its precursors, and routine parameters of the SAH group over the observation period. Correlation between laboratory data over all 15 days combined. Correlation coefficients and significance were calculated using Spearman’s rho test. **Correlation is significant at the 0.01 level (2-tailed). *Correlation is significant at the 0.05 level (2-tailed). The TMAO indicates trimethylamine N-oxide; SAH, subarachnoid hemorrhage; RBC, red blood cell; eGFR, estimated glomerular filtration rate; aPTT, activated partial thromboplastin time
TMAO and precursors at recovery or improvement
Plasma TMAO levels in the SAH group were significantly lower at recovery or the end of the observation period (improvement) than the control group at baseline (1.9 versus 2.9 μmol/L). Furthermore, plasma betaine levels were also significantly lower in the SAH group at this time point. In contrast, plasma choline levels were not significantly lower in the SAH group, and plasma carnitine levels were even not significantly higher compared to the control group (Supplemental Table 6).
Discussion
Trimethylamine N-oxide is often associated with various diseases, such as atherosclerosis, diabetes, cancer, or stroke [14]. Since TMAO most probably affects the platelet responsiveness in humans, and thrombotic events may play a substantial role in the pathogenesis of SAH and its complications, investigations seem reasonable to clarify the role of TMAO, also as a biomarker, in these thrombotic processes [5, 53].
In our study, we found decreased plasma TMAO levels in patients with SAH compared to non-SAH controls. At recovery or the end of the observation period, we continued to find significantly lower plasma TMAO levels in the SAH group. Plasma TMAO levels of our control group were comparable to levels in a previous study with a central European cohort [23]. Additionally, hypertension occurred more often in the SAH group of our study. Hypertension is a typical risk factor for SAH but has also been associated with higher plasma TMAO levels previously [15, 41]. No other differences in demographics could be found. Mechanisms of action of TMAO include promotion of vascular inflammation, an important pathomechanism of cerebral aneurysms, and TMAO may even lead to disruption of the blood–brain barrier [3, 21, 51]. We hypothesized that patients with higher levels of TMAO would be at higher risk for SAH a priori because of the aforementioned mechanisms and that our patient group would therefore have higher TMAO levels on admission. In a hypertensive population, this effect should have been even greater.
Increased and decreased plasma TMAO levels have been reported for stroke patients, previously [11, 45, 49]. Schneider et al. could show that increased plasma TMAO levels on admission are followed by a decrease within the following 2 days [38]. This may be an explanation for contrary results. We could not find any characteristic course or significant changes in plasma TMAO levels after SAH, so TMAO levels remained decreased. Studies on TMAO in stroke patients are based mainly on ischemic stroke patients in mixed cohorts and should only be an indication for further research on TMAO in SAH patients. Risk factors, prediction, genesis, and management differ distinctively between ischemic and hemorrhagic stroke [1, 37, 46]. To the best of our knowledge, TMAO has not been investigated in patients with SAH. Hence, results should not only be interpreted based on recent findings. These differences could explain a lower plasma TMAO level in SAH patients compared to stroke patients in other trials.
One explanation for decreased TMAO levels in the SAH group could also be the so-called gut-brain axis, which refers to a “bidirectional communication network” between the nervous system and intestine [47]. We ingest TMAO mainly by eating fish, eggs, and meat products. Its precursors, e.g., betaine, carnitine, and choline, are metabolized by certain bacterial species of the gut microbiome. The product, trimethylamine (TMA), is incorporated via the intestine, transported to the liver, and metabolized by flavin-containing monooxygenases to TMAO [2, 6, 24, 32, 50]. Decreased plasma TMAO levels may result from acute brain damage and subsequent gut dysbiosis [4]. Patients with SAH typically experience sympathetic activation and a so-called catecholamine surge which could lead to downregulation of the immune system in the gut. Thereupon, the composition of the microbiome changes [18, 20]. Beneficial microbiota become increasingly deficient. The resulting decrease in TMAO levels has already been observed in patients with stroke or transient ischemic attack [49]. Apparently, the microspecies responsible for TMA production also perish or reduce production.
Worsened renal function is also reported to have a major impact on TMAO levels and explains higher plasma TMAO levels [31]. Although lower kidney function is reported in stroke patients, eGFR in the SAH group on admission was significantly higher (99 mL/min/1.73 m2) compared to the control group (90 mL/min/1.73 m2), and higher than the reference threshold of 90 mL/min/1.73 m2 in both groups [52]. The eGFR of the SAH group remained higher than the reference threshold for the entire observational period. The informational value of the eGFR for kidney function is distinctively lower, with an eGFR higher than 60 mL/min/1.73 m2 [39]. Accordingly, in our trial, we could hardly trace differences in plasma TMAO levels to the kidney function.
We did not find evidence in this trial for an association between the plasma TMAO and the clinical severity of SAH, but we did find hints of a negative correlation. Further investigations with higher numbers of cases will be necessary to prove such an association.
We expected higher TMAO levels in patients who develop DCI. DCI seems to be caused by an interplay of vascular inflammation and activation of the coagulation system, among other factors. This leads to vasospasm in vessels, from large arteries to arterioles, and microthrombus formation in small vessels [5, 44]. These effects could be exacerbated by the inflammatory and thrombotic potential of TMAO and could be more likely to lead to the development of DCI in patients with higher TMAO levels during the course of SAH. We did not find the expected difference between DCI and non-DCI cases and cannot support this hypothesis in our study. A reason for this could be the small number of 9 DCI cases.
Our results confirm the appearance of TMAO in the CSF and a correlation between plasma and CSF TMAO levels had already been reported. The correlation between the CSF TMAO and the plasma TMAO may be caused by BCB disturbances, but we did not find any correlation between QALB and QTMAO. BCB disorders typically occur after SAH, due to the destruction of endothelial cells and the disruption of tight junctions [25]. The BCB does not seem to be more permeable for TMAO, even if it is more permeable for albumin. Hence, we cannot support the conclusion made by a recent trial investigating CSF from 290 lumbar punctures [9]. The authors found a positive correlation between CSF TMAO and serum TMAO and an influence of the CSF/serum albumin ratio on the CSF/serum TMAO ratio, concluding that TMAO may cross the BCB via passive diffusion. Our results indicate that TMAO is transported over the BCB via active transport. CSF TMAO levels could potentially be a biomarker for or be associated with the genesis of central nervous diseases of any kind. Further investigations with a higher sample count are mandatory to confirm these observations.
The increase in precursors on the 5th to 7th day after SAH may be a sign of gut recovery. The missing increase of TMAO may be due to the length of the observation period. We also hypothesized that patients with thrombotic complications, such as DCI, would have an increase in TMAO after SAH. Although the pathogenesis of DCI can be attributed to vasospasm, the development of microvascular thrombi also seems to play an important role. It is well known that TMAO is associated with thrombotic events and atherosclerosis [17]. Zhu et al. revealed the possible promoting influence of TMAO on platelet responsiveness [53]. The pathologies remain largely unclear and mostly relate to cardiovascular disease. Unfortunately, there were too few cases of DCI in our study group to detect abnormalities in TMAO levels.
Interestingly, the platelet count started to increase simultaneously on the 5th day. A reactive thrombocytosis has already been described after SAH and could explain the increase [19]. A protective influence of carnitine on the platelet metabolism and function in stored platelet concentrates had also been found [7]. So far, the focus lies on the effect of TMAO and its precursors on platelet activity, but perhaps more attention should be paid to their effect on platelet metabolism, especially when evaluating the influence of TMAO on thrombus formation and DCI in SAH patients [5, 53].
Limitations of this study are the small count of SAH patients and the decreasing sample count during the observation period in the intensive care unit. Additionally, we were not able to determine the exact time point of hemorrhage and, therefore, could not integrate the time between the onset of symptoms and the first TMAO measurement into our analysis.
Conclusions
Our results do not confirm our hypothesis that increased TMAO levels occur in patients with SAH or are related to its complications. Unexpectedly, plasma TMAO levels are decreased in SAH patients. The detection of TMAO in the CSF emphasizes a possible role of TMAO in central nervous processes. An impact of TMAO, or its absence, on patients with SAH or a role as a biomarker for the appearance of SAH and its complications would advance the understanding of this disease and could improve their management in the hospital. The exact effects and characteristics of TMAO in the human body should remain the focus of research. Further research with a larger number of SAH and DCI cases and different ethnicities is needed. Moreover, an analysis of the patients’ feces and the microorganisms contained should be added to the next investigations. The acquisition of TMAO into standardized biomarker patterns could, therefore, be a helpful instrument.
Data availability
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to data privacy.
References
Andersen KK, Olsen TS, Dehlendorff C, Kammersgaard LP (2009) Hemorrhagic and ischemic strokes compared: stroke severity, mortality, and risk factors. Stroke 40:2068–2072. https://doi.org/10.1161/STROKEAHA.108.540112
Arias N, Arboleya S, Allison J, Kaliszewska A, Higarza SG, Gueimonde M, Arias JL (2020) The relationship between choline bioavailability from diet, intestinal microbiota composition, and its modulation of human diseases. Nutrients 12. https://doi.org/10.3390/nu12082340
Chalouhi N, Hoh BL, Hasan D (2013) Review of cerebral aneurysm formation, growth, and rupture. Stroke 44:3613–3622. https://doi.org/10.1161/STROKEAHA.113.002390
Cho J, Park YJ, Gonzales-Portillo B, Saft M, Cozene B, Sadanandan N, Borlongan CV (2021) Gut dysbiosis in stroke and its implications on Alzheimer’s disease-like cognitive dysfunction. CNS Neurosci Ther. https://doi.org/10.1111/cns.13613
Clarke JV, Suggs JM, Diwan D, Lee JV, Lipsey K, Vellimana AK, Zipfel GJ (2020) Microvascular platelet aggregation and thrombosis after subarachnoid hemorrhage: a review and synthesis. J Cereb Blood Flow Metab 40:1565–1575. https://doi.org/10.1177/0271678X20921974
Coutinho-Wolino KS, Cardozo LFM, de Oliveira Leal V, Mafra D, Stockler-Pinto MB (2021) Can diet modulate trimethylamine N-oxide (TMAO) production? What do we know so far? Eur J Nutr. https://doi.org/10.1007/s00394-021-02491-6
Deyhim MR, Mesbah-Namin SA, Yari F, Taghikhani M, Amirizadeh N (2015) L-carnitine effectively improves the metabolism and quality of platelet concentrates during storage. Ann Hematol 94:671–680. https://doi.org/10.1007/s00277-014-2243-5
Emonds JJ, Ringel C, Reinicke M, Müller D, von Eckardstein A, Meixensberger J, Ceglarek U, Gaudl A (2022) Influence of trimethylamine N-Oxide on platelet activation. Nutrients 14:3261. https://doi.org/10.3390/nu14163261
Enko D, Zelzer S, Niedrist T, Holasek S, Baranyi A, Schnedl WJ, Herrmann M, Meinitzer A (2020) Assessment of trimethylamine-N-oxide at the blood-cerebrospinal fluid barrier: results from 290 lumbar punctures. EXCLI J 19:1275–1281. https://doi.org/10.17179/excli2020-2763
Etminan N, Chang H-S, Hackenberg K, de Rooij NK, Vergouwen MDI, Rinkel GJE, Algra A (2019) Worldwide incidence of aneurysmal subarachnoid hemorrhage according to region, time period, blood pressure, and smoking prevalence in the population: a systematic review and meta-analysis. JAMA Neurol 76:588–597. https://doi.org/10.1001/jamaneurol.2019.0006
Farhangi MA, Vajdi M, Asghari-Jafarabadi M (2020) Gut microbiota-associated metabolite trimethylamine N-Oxide and the risk of stroke: a systematic review and dose-response meta-analysis. Nutr J 19:76. https://doi.org/10.1186/s12937-020-00592-2
Feigin VL, Lawes CMM, Bennett DA, Barker-Collo SL, Parag V (2009) Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol 8:355–369. https://doi.org/10.1016/S1474-4422(09)70025-0
Fisher CM, Kistler JP, Davis JM (1980) Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 6:1–9. https://doi.org/10.1227/00006123-198001000-00001
Gatarek P, Kaluzna-Czaplinska J (2021) Trimethylamine N-oxide (TMAO) in human health. EXCLI J 20:301–319. https://doi.org/10.17179/excli2020-3239
Ge X, Zheng L, Zhuang R, Yu P, Xu Z, Liu G, Xi X, Zhou X, Fan H (2020) The gut microbial metabolite trimethylamine N-oxide and hypertension risk: a systematic review and dose-response meta-analysis. Adv Nutr 11:66–76. https://doi.org/10.1093/advances/nmz064
Gerner ST, Reichl J, Custal C, Brandner S, Eyüpoglu IY, Lücking H, Hölter P, Kallmünzer B, Huttner HB (2020) Long-term complications and influence on outcome in patients surviving spontaneous subarachnoid hemorrhage. CED 49:307–315. https://doi.org/10.1159/000508577
Guasti L, Galliazzo S, Molaro M, Visconti E, Pennella B, Gaudio GV, Lupi A, Grandi AM, Squizzato A (2021) TMAO as a biomarker of cardiovascular events: a systematic review and meta-analysis. Intern Emerg Med 16:201–207. https://doi.org/10.1007/s11739-020-02470-5
Hasegawa Y, Uchikawa H, Kajiwara S, Morioka M (2022) Central sympathetic nerve activation in subarachnoid hemorrhage. J Neurochem 160:34–50. https://doi.org/10.1111/jnc.15511
Hirashima Y, Hamada H, Kurimoto M, Origasa H, Endo S (2005) Decrease in platelet count as an independent risk factor for symptomatic vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 102:882–887. https://doi.org/10.3171/jns.2005.102.5.0882
Huang Q, Xia J (2021) Influence of the gut microbiome on inflammatory and immune response after stroke. Neurol Sci 42:4937–4951. https://doi.org/10.1007/s10072-021-05603-6
Janeiro MH, Ramírez MJ, Milagro FI, Martínez JA, Solas M (2018) Implication of trimethylamine N-oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients 10. https://doi.org/10.3390/nu10101398
Kirsch SH, Herrmann W, Rabagny Y, Obeid R (2010) Quantification of acetylcholine, choline, betaine, and dimethylglycine in human plasma and urine using stable-isotope dilution ultra performance liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 878:3338–3344. https://doi.org/10.1016/j.jchromb.2010.10.016
Krüger R, Merz B, Rist MJ, Ferrario PG, Bub A, Kulling SE, Watzl B (2017) Associations of current diet with plasma and urine TMAO in the KarMeN study: direct and indirect contributions. Mol Nutr Food Res 61. https://doi.org/10.1002/mnfr.201700363
Li X, Hong J, Wang Y, Pei M, Wang L, Gong Z (2021) Trimethylamine-N-oxide pathway: a potential target for the treatment of MAFLD. Front Mol Biosci 8:733507. https://doi.org/10.3389/fmolb.2021.733507
Li Y, Wu P, Bihl JC, Shi H (2020) Underlying mechanisms and potential therapeutic molecular targets in blood-brain barrier disruption after subarachnoid hemorrhage. Curr Neuropharmacol 18:1168–1179. https://doi.org/10.2174/1570159X18666200106154203
Macdonald RL, Schweizer TA (2017) Spontaneous subarachnoid haemorrhage. The Lancet 389:655–666. https://doi.org/10.1016/S0140-6736(16)30668-7
Neifert SN, Chapman EK, Martini ML, Shuman WH, Schupper AJ, Oermann EK, Mocco J, Macdonald RL (2020) Aneurysmal subarachnoid hemorrhage: the last decade. Transl Stroke Res. https://doi.org/10.1007/s12975-020-00867-0
Nieuwkamp DJ, Setz LE, Algra A, Linn FHH, de Rooij NK, Rinkel GJE (2009) Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol 8:635–642. https://doi.org/10.1016/S1474-4422(09)70126-7
Ocque AJ, Stubbs JR, Nolin TD (2015) Development and validation of a simple UHPLC-MS/MS method for the simultaneous determination of trimethylamine N-oxide, choline, and betaine in human plasma and urine. J Pharm Biomed Anal 109:128–135. https://doi.org/10.1016/j.jpba.2015.02.040
Ottiger M, Nickler M, Steuer C, Odermatt J, Huber A, Christ-Crain M, Henzen C, Hoess C, Thomann R, Zimmerli W et al (2016) Trimethylamine-N-oxide (TMAO) predicts fatal outcomes in community-acquired pneumonia patients without evident coronary artery disease. Eur J Intern Med 36:67–73. https://doi.org/10.1016/j.ejim.2016.08.017
Pelletier CC, Croyal M, Ene L, Aguesse A, Billon-Crossouard S, Krempf M, Lemoine S, Guebre-Egziabher F, Juillard L, Soulage CO (2019) Elevation of trimethylamine-N-oxide in chronic kidney disease: contribution of decreased glomerular filtration rate. Toxins (Basel) 11. https://doi.org/10.3390/toxins11110635
Rath S, Rud T, Pieper DH, Vital M (2019) Potential TMA-producing bacteria are ubiquitously found in mammalia. Front Microbiol 10:2966. https://doi.org/10.3389/fmicb.2019.02966
Reiber H (2016) Knowledge-base for interpretation of cerebrospinal fluid data patterns. Essentials in neurology and psychiatry. Arq Neuropsiquiatr 74:501–512. https://doi.org/10.1590/0004-282X20160066
Report of World Federation of Neurological Surgeons Committee on a Universal Subarachnoid Hemorrhage Grading Scale (1988) J Neurosurg. 68, 985–986. https://doi.org/10.3171/jns.1988.68.6.0985
Rexidamu M, Li H, Jin H, Huang J (2019) Serum levels of trimethylamine-N-oxide in patients with ischemic stroke. Biosci Rep, 39, 10.1042/BSR20190515
Ringel C, Dittrich J, Gaudl A, Schellong P, Beuchel CF, Baber R, Beutner F, Teren A, Engel C, Wirkner K et al (2021) Association of plasma trimethylamine N-oxide levels with atherosclerotic cardiovascular disease and factors of the metabolic syndrome. Atherosclerosis 335:62–67. https://doi.org/10.1016/j.atherosclerosis.2021.09.026
Sacco RL, Kasner SE, Broderick JP, Caplan LR, Connors JJB, Culebras A, Elkind MSV, George MG, Hamdan AD, Higashida RT et al (2013) An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 44:2064–2089. https://doi.org/10.1161/STR.0b013e318296aeca
Schneider C, Okun JG, Schwarz KV, Hauke J, Zorn M, Nürnberg C, Ungerer M, Ringleb PA, Mundiyanapurath S (2020) Trimethylamine-N-oxide (TMAO) is elevated in the acute phase after ischemic stroke and decreases within the first days. Eur J Neurol. https://doi.org/10.1111/ene.14253
Stevens LA, Coresh J, Greene T, Levey AS (2006) Assessing kidney function–measured and estimated glomerular filtration rate. N Engl J Med 354:2473–2483. https://doi.org/10.1056/NEJMra054415
Subramaniam S, Fletcher C (2018) Trimethylamine N-oxide: breathe new life. Br J Pharmacol 175:1344–1353. https://doi.org/10.1111/bph.13959
Sundström J, Söderholm M, Söderberg S, Alfredsson L, Andersson M, Bellocco R, Björck M, Broberg P, Eriksson M, Eriksson M et al (2019) Risk factors for subarachnoid haemorrhage: a nationwide cohort of 950 000 adults. Int J Epidemiol 48:2018–2025. https://doi.org/10.1093/ije/dyz163
Suzuki T, Heaney LM, Bhandari SS, Jones DJL, Ng LL (2016) Trimethylamine N-oxide and prognosis in acute heart failure. Heart 102:841–848. https://doi.org/10.1136/heartjnl-2015-308826
Suzuki T, Heaney LM, Jones DJL, Ng LL (2017) Trimethylamine N-oxide and risk stratification after acute myocardial infarction. Clin Chem 63:420–428. https://doi.org/10.1373/clinchem.2016.264853
Suzuki H, Kanamaru H, Kawakita F, Asada R, Fujimoto M, Shiba M (2021) Cerebrovascular pathophysiology of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Histol Histopathol 36:143–158. https://doi.org/10.14670/HH-18-253
Tan C, Wang H, Gao X, Xu R, Zeng X, Cui Z, Zhu J, Wu Q, Xia G, Zhou H et al (2020) Dynamic changes and prognostic value of gut microbiota-dependent trimethylamine-N-oxide in acute ischemic stroke. Front Neurol 11:29. https://doi.org/10.3389/fneur.2020.00029
Tsai C-F, Anderson N, Thomas B, Sudlow CLM (2016) Comparing risk factor profiles between intracerebral hemorrhage and ischemic stroke in chinese and white populations: systematic review and meta-analysis. PLoS ONE 11:e0151743. https://doi.org/10.1371/journal.pone.0151743
Wang H-X, Wang Y-P (2016) Gut Microbiota-brain Axis. Chin Med J 129:2373–2380. https://doi.org/10.4103/0366-6999.190667
Xiong Y, Zhao Y-Y, Goruk S, Oilund K, Field CJ, Jacobs RL, Curtis JM (2012) Validation of an LC-MS/MS method for the quantification of choline-related compounds and phospholipids in foods and tissues. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 911, 170–179. https://doi.org/10.1016/j.jchromb.2012.10.038
Yin J, Liao S-X, He Y, Wang S, Xia G-H, Liu F-T, Zhu J-J, You C, Chen Q, Zhou L et al. (2015) Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J Am Heart Assoc 4. https://doi.org/10.1161/JAHA.115.002699
Zeisel SH, Warrier M (2017) Trimethylamine N-oxide, the microbiome, and heart and kidney disease. Annu Rev Nutr 37:157–181. https://doi.org/10.1146/annurev-nutr-071816-064732
Zhang Y, Wang Y, Ke B, Du J (2021) TMAO: how gut microbiota contributes to heart failure. Transl Res 228:109–125. https://doi.org/10.1016/j.trsl.2020.08.007
Zhao Q, Yan T, Chopp M, Venkat P, Chen J (2020) Brain-kidney interaction: renal dysfunction following ischemic stroke. J Cereb Blood Flow Metab 40:246–262. https://doi.org/10.1177/0271678X19890931
Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M et al (2016) Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 165:111–124. https://doi.org/10.1016/j.cell.2016.02.011
Acknowledgements
We thank the study department of the Institute of Laboratory Medicine, Clinical Chemistry and Molecular Diagnostics, University of Leipzig Medical Center (Leipzig, Germany), and the personnel of the interdisciplinary surgical intensive care unit, University of Leipzig Medical Center (Leipzig, Germany), for their considerable support of this study.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was funded by a grant from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 209933838 – Collaborative Research Center SFB1052 “Obesity Mechanisms,” to U.C. (SFB-1052/A9). Julian Emonds received a doctoral scholarship from the Faculty of Medicine, University of Leipzig. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results. The publication was supported by Open Access Publishing Fund of Leipzig University.
Author information
Authors and Affiliations
Contributions
Conceptualization: Julian Josef Emonds, Felix Arlt, Alexander Gaudl, Jürgen Meixensberger, and Uta Ceglarek. Methodology: Julian Josef Emonds, Felix Arlt, Alexander Gaudl, Jürgen Meixensberger, and Uta Ceglarek. Validation: Alexander Gaudl, Madlen Reinicke, and Uta Ceglarek. Formal analysis: Julian Josef Emonds, Jürgen Meixensberger, and Uta Ceglarek. Investigation: Julian Josef Emonds, Felix Arlt, Jürgen Meixensberger, Dirk Lindner, Sven Laudi, Alexander Gaudl, Madlen Reinicke, and Uta Ceglarek. Resources: Sven Laudi, Jürgen Meixensberger, and Uta Ceglarek. Data curation: Julian Josef Emonds. Writing—original draft preparation: Julian Josef Emonds, Alexander Gaudl, Madlen Reinicke, Mitja Heinemann, Felix Arlt, and Jürgen Meixensberger. Writing—review and editing: Julian Josef Emonds, Felix Arlt, Alexander Gaudl, Madlen Reinicke, Mitja Heinemann, Dirk Lindner, Sven Laudi, Uta Ceglarek, and Jürgen Meixensberger. Visualization: Julian Josef Emonds. Supervision: Jürgen Meixensberger and Uta Ceglarek. Project administration: Julian Josef Emonds, Felix Arlt, Alexander Gaudl, Jürgen Meixensberger, and Uta Ceglarek. Funding acquisition: Jürgen Meixensberger and Uta Ceglarek. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Medical Faculty of the University of Leipzig (ethical approval 236/18-ek; date of approval July 31, 2018).
Informed consent
Written informed consent was obtained from all participants or their legal representatives.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Comments
The authors investigated an exciting hypothesis. Delayed cerebral ischaemia ascribed to vasospasm in aneurysmal subarachnoid is one of the most important causes of unfortunate outcomes in this disease. Despite intensive research and the availability of approved drug treatment (nimodipine), many pts cannot be rescued. The authors considered that microbiome-derived trimethylamine N-oxide (TMAO) should enhance platelet responsiveness and be thrombophilic, leading to vasospasm. They performed a prospective observational study to evaluate TMAO in patients with subarachnoid haemorrhage (SAH) and compared it with a control group. The secondary aim was to measure TMAO in the cerebrospinal fluid (CSF) from these patients.
No association between the plasma TMAO and the clinical severity of SAHs were found; however, there were hints of a negative correlation.
They did not find the expected difference between DCI and non-DCI cases. The results do not confirm their hypothesis that increased TMAO levels would occur in patients with SAH or are related to its complications. Unexpectedly, plasma TMAO levels were decreased in SAH patients, which may hint toward an influence of the gut-brain axis. They conclude that detecting TMAO in the CSF may emphasize the possible role of TMAO in central nervous processes.
Unfortunately, conclusions cannot be drawn due to the low number of cases and due to a lack of data considering the microbiome. However, further research with more SAH and DCI cases and different ethnicities would help answer these questions. This study does not exclude that acquisition of TMAO into standardized biomarker patterns could not be a helpful instrument in the future.
Tamas Peter Dozi
Pecs, Hungary
This article is part of the Topical Collection on Neurosurgical intensive care
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Emonds, J.J., Arlt, F., Gaudl, A. et al. Trimethylamine N-oxide (TMAO) in patients with subarachnoid hemorrhage: a prospective observational study. Acta Neurochir 165, 1277–1287 (2023). https://doi.org/10.1007/s00701-022-05485-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-022-05485-3