
Abstract
Background
Fatigue is a common and disabling sequel after aneurysmal subarachnoid hemorrhage (aSAH). At present, prevalence estimates of post-aSAH fatigue in the chronic phase are scarce and vary greatly. Factors from the acute phase of aSAH have hitherto barely been associated with post-aSAH fatigue in the chronic phase.
Methods
Prospective study assessing prevalence of fatigue using the Fatigue Severity Scale (FSS) in patients who were living independently 1 to 7 years after aSAH. We compared demographic, medical, and radiological variables from the acute phase of aSAH between patients with and without fatigue (FSS ≥ 4 versus < 4) and searched for predictors of fatigue among these variables applying univariable and multivariable regression analyses.
Results
Of 726 patients treated for aSAH in the period between January 2012 and December 2017, 356 patients completed the assessment. The mean FSS score was 4.7 ± 1.7, and fatigue was present in 69.7%. The frequency of patients with fatigue did not decline significantly over time. Univariable analysis identified nicotine use, loss of consciousness at ictus (LOCi), rebleed prior to aneurysm repair, reduced consciousness to Glasgow Coma Scale (GCS) < 14, large amounts of subarachnoid blood, the presence of acute hydrocephalus, and severe vasospasm as factors that were significantly associated with fatigue. In multivariable analysis, nicotine use, reduced GCS, and severe vasospasm were independent predictors that all more than doubled the risk to develop post-aSAH fatigue.
Conclusions
Fatigue is a frequent sequel persisting several years after aSAH. Nicotine use, reduced consciousness at admission, and severe vasospasm are independent predictors of fatigue from the acute phase of aSAH. We propose inflammatory cytokines causing dopamine imbalance to be a common denominator for post-aSAH fatigue and the presently identified predictors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fatigue can be characterized by “a feeling of lack of energy, weariness, and aversion to effort” [32]. Currently there is no agreed-upon definition or operationalization of fatigue, partly a result of its complex and multidimensional nature. Fatigue is the most prominent sequel in a cluster of residual symptoms after aneurysmal subarachnoid hemorrhage (aSAH) [34]. Although being highly frequent in the early phase [36, 41], post-aSAH fatigue is also present several years after the ictus [7, 44] and it is a debilitating symptom that has a significant impact upon quality of life and the ability to return to work [38, 43]. Estimates of fatigue prevalence after aSAH vary between 31 and 90 % [28]. This large variability may be related to heterogeneity in study population, timing of assessment after the hemorrhage and methods of assessment for fatigue.
Since fatigue is common in many neurological disorders [9], there may be a range of different underlying neurobiological mechanisms. The pathophysiology of fatigue after ischemic and hemorrhagic stroke remains unclear [11] and the knowledge of aSAH-related factors associated with post-aSAH fatigue is even more limited. Only a small number of studies have examined multiple clinical predictors and their relationship with post-aSAH fatigue [7, 26, 37, 41]. In combination with relative small sample sizes in previous studies, factors associated with post-aSAH fatigue have not been comprehensively evaluated.
Subjects and methods
Subjects
The current study was part of the pre-screening for a double-blind, randomized, placebo-controlled study investigating the effect of OSU6162 in the treatment of fatigue and other neuropsychological sequelae after aSAH (http://www.clinicaltrials.gov. Unique identifier: NCT03209830) approved by Health Research Ethics (REC, reference: 2016/2214). Patients (≥ 18 years) that had suffered aSAH at least 1 year earlier and who were treated for their aSAH at our department between January 2012 and December 2017 were contacted. Patients who agreed to participate were interviewed by phone and gave an oral informed consent. Patients living permanently in an institution and those not speaking adequately Norwegian were excluded. The data collection protocol was approved by the institutional review board. The study data that support the findings of this study are available from the corresponding author on reasonable request.
Measures
Fatigue
Fatigue in the chronic phase (≥ 1 year) after aSAH was assessed using the Norwegian version of the Fatigue Severity Scale (FSS) [27, 30] which consists of nine statements about the impact of fatigue on daily life; each statement is scored on a 7-point Likert scale, ranging from 1 (strongly disagree) to 7 (strongly agree). The total FSS score is the mean of the item scores. A mean score of 4 or more is considered outside the range of healthy controls [27] and indicates a moderate to high impact of fatigue on daily living.
Predictors
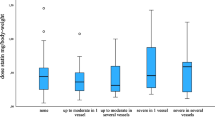
Data on demographics (age and sex), medical condition at admission, and clinical course during the acute phase of aSAH were obtained from our institutional quality registry. We registered nicotine use at the time of ictus, if there was an abrupt LOCi including seizures, rebleed before aneurysm repair, clinical grade just prior to aneurysm repair or prior to intubation in patients admitted intubated (Hunt and Hess Scale [HH] [21] and Glasgow Coma Scale (GCS) [23]), aneurysm location, and the method of aneurysm repair. From the diagnostic computed tomography (CT), we scored the presence of intraparenchymal hemorrhage, the amount of subarachnoid blood (modified Fisher grade [18]), and the amount of intraventricular blood (modified LeRoux score [29], where no intraventricular blood was scored as “0”). Furthermore, we registered if the patient was treated for acute hydrocephalus (need of external drainage of CSF), or for chronic hydrocephalus (implanted shunt). All patients underwent a CT/CT angiography (CTA) on the first day after aneurysm repair and on day 5 in intubated patients and on day 7 in awake patients. Transcranial Doppler ultrasonography (TCD) was performed from day 4 and at regular intervals. We scored the presence of severe vasospasm if CT angiography scans showed a > 50% diameter reduction in one or several vessels and/or TCD showing a Lindegaard ratio [31] > 6. The acquisition of a new cerebral infarction, including those that were procedure-related, was evaluated from CT scans performed during the primary hospital stay or from magnetic resonance imaging (MRI) if performed.
Statistical analysis
Patient characteristics are presented as mean values with standard deviation (SD) or proportions. Differences in continuous variables between groups were tested using independent sample t test or one-way ANOVA. The chi-square test for contingency tables was used to detect associations between categorical variables. To evaluate the frequency of fatigue across time intervals in years since ictus, a Mantel-Haenszel test for trend was performed. Univariable and multivariable logistic regression analyses were performed to identify possible predictors of fatigue (FSS ≥ 4): LOCi (yes or no), rebleed (yes or no), Hunt and Hess scale (1–3 or 4–5), modified Fisher scale (0–2 or 3–4), modified LeRoux score (0–5 or 6–16), severe vasospasm (yes or no), acute hydrocephalus (yes or no), chronic hydrocephalus (yes or no), intracerebral hemorrhage (ICH) (yes or no), and cerebral infarction (yes or no) were dichotomized. Aneurysm location, GCS, and nicotine use were entered by using 3 dummy variables. Any variable with p < 0.05 in the univariable analysis was considered a candidate for the multivariable model. Subsequent multivariable logistic regression analyses with manual backward elimination were performed. The associations between potential predictors and fatigue were quantified by odds ratio (OR) with 95% confidence interval (CI). Multivariable analyses were preceded by estimation of correlation between predictors. Two-tailed p values of less than 5% were considered statistically significant. All statistical analyses were performed using the IBM SPSS statistics version 25.0 (IBM SPSS Inc., Armonk, NY: IBM Corp).
Results
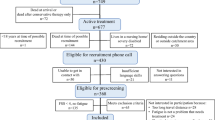
A total of 726 patients were admitted for non-traumatic aSAH between January 2012 and December 2017. Figure 1 shows the flow chart of eligible and included patients. A total of 655 patients received active treatment. In this group, 138 died, 69 were severely disabled and/or resided in a nursing home, 30 were living outside Health Region South East, and one was younger than 18 years. Therefore, 417 were eligible for telephone interview, of whom 356 (85%) participated in the study. We excluded 21 patients with insufficient skills in Norwegian, 30 patients were unable to be traced or did not answer, and 10 patients declined participation. The characteristics of the 356 included patients are displayed in Table 1.

Patient enrollment
Prevalence and duration of fatigue
The mean FSS score was 4.7 (SD, 1.7) and fatigue (FSS ≥ 4) was present in 248 patients (69.7%) (Table 1). Figure 2 shows the prevalence of fatigue in relation to time passed since the aSAH. From 1 and up to 7 years after the ictus, fatigue was present in 74%, 74%, 61%, 77%, 65%, and 60%, respectively. Even though there seemed to be a tendency towards decline of fatigue as a function of time, the Mantel-Haenszel test for trend demonstrated that fatigue was stable over time (p 0.057).

Percentage of patients with fatigue in relation to months passed since ictus
Predictors of fatigue
Results of the univariable analyses on predictors linked to the acute phase of aSAH (demographic, medical, and radiological data) are presented in Table 2. Nicotine use (OR 2.49, 95% CI 1.50–4.14; p < 0.001), reduced consciousness to GCS 13–9 (OR 2.46, 95% CI 1.21–4.98; p = 0.013) and GCS 8–3 (OR 2.30, 95% CI 1.16–4.56; p = 0.017), larger amount of subarachnoid blood (modified Fisher scales 3 and 4: OR 1.86, 95% CI 1.18–2.94; p = 0.008), LOCi (OR 2.04, 95% CI 1.26–3.30; p = 0.004), rebleed before aneurysm repair (OR 3.05, 95% CI 1.04–8.95; p = 0.043), severe vasospasm (OR 2.49, 95% CI 1.21–5.12; p = 0.013), and acute hydrocephalus (OR 2.08, 95% CI 1.30–3.32; p = 0.002) were identified as statistically significant predictors of fatigue. The mean FSS was significantly lower in never nicotine users (4.20 ± 1.80) than in former users (4.65 ± 1.55) and current users (4.91 ± 1.61) (p = 0.002; Fig. 3, left). Likewise, the mean FSS was significantly lower in patients with GCS 15–14 (4.48 ± 1.79) than in those with GCS 13–9 (4.91 ± 1.44) and those with GCS 8–3 (5.08 ± 1.40) (p = 0.020; Fig. 3, right).

Mean Fatigue Severity Score (FSS) versus nicotine use (left) and Glasgow Coma Score (right)
The final multivariable regression model is presented in Table 2, right column, and identified nicotine use, GCS < 14, and severe vasospasm as independent predictors which approximately doubled the risk to develop fatigue after aSAH: nicotine use (OR 2.10, 95% CI 1.31–3.39; p = 0.002) as compared with former and never nicotine use, patients in GCS 13–9 (OR 2.49, 95% CI 1.21–5.14; p = 0.014) and GCS 8–3 (OR 2.13, 95% CI 1.03–4.38; p = 0.040) as compared with patients in GCS 15–14, and patients with severe vasospasm (OR 2.30, 95% CI 1.10–4.82; p = 0.028) as compared with patients with less severe or no vasospasm.
The interwoven relationship of clinical variables related to aSAH is illustrated in Fig. 4. We focused on the presently identified predictors of post-aSAH fatigue and how they possibly may culminate in processes leading to or facilitating the development of fatigue.

Relationship of variables of aneurysmal hemorrhage, which culminate in processes attributable to the development of fatigue. Black boxes indicate independent predictors of fatigue in the present study. GCS, Glasgow Coma Score [23]; ATP, adenosine triphosphate
Discussion
The core finding in the present study was that the prevalence of fatigue in the chronic phase after aSAH was 69.7% and remained stable over at least 1 to 7 years after the hemorrhage. Nicotine use, reduced consciousness at admission, and severe vasospasm in the acute phase were independent predictors of fatigue.
Prevalence of post-aSAH fatigue
Our prevalence of 70% is consistent with previous studies that assessed the prevalence of fatigue by means of the FSS questionnaire in patients surviving aSAH [28]. However, other studies that used single questions to assess fatigue reported a much more widespread prevalence [28]. The variance in prevalence is not surprising since there is no “gold standard” or objective measure of fatigue, and the lack of consensus regarding the definition of fatigue is therefore a challenge for allowing consistent measures [13]. Self-report measures of fatigue have major limitations, especially questionnaires that fail to regard fatigue as a multidimensional state with cognitive and emotional/psychological components. Another factor contributing to varying results regarding prevalence of fatigue is low sample size. To the best of our knowledge, the 356 aSAH patients we presently examined comprise by far the largest cohort investigated on this topic to date.
Timely evolvement of post-aSAH fatigue
Previous longitudinal studies show a consistency in fatigue levels over time [3, 19, 26, 39], where no improvement of fatigue was found in studies with follow-up assessment up to 4 years after the ictus [3, 19]. In line with our findings, other cross-sectional studies [6, 7, 43] have shown a high prevalence of fatigue (47–66%) up to 10 years after the ictus. A systematic review by Kutlubaev et al. [28] reported a higher frequency of fatigue less than 1 year compared with more than 1 year after the ictus (73.6% and 50.7%, respectively), which they postulate can be explained by different mechanisms driving fatigue as a function of time since ictus. On the other hand, in a meta-analysis by Cumming et al. [10], a marked between-study variability in the estimates of fatigue prevalence was not accounted for by timing of assessment after stroke. The authors therefore concluded fatigue to be persistent across time. Hitherto, there are insufficient data to draw any firm conclusions regarding time course; however, there are strong indications that fatigue remains relatively stable after the acute phase. Our data were acquired in patients where the hemorrhage was at least 1 year ago and support the notion that fatigue beyond 1 year is best understood as a chronic condition.
The dopamine imbalance hypothesis
Not only the definition and quantification of fatigue is challenging, also the underlying biomedical mechanisms remain unclear. Since central fatigue [8] is seen in a wide specter of diseases that share some kind of proinflammatory mechanism, the hypothesis of cytokine-induced inflammation-mediated fatigue has emerged [4, 25, 40]. Inflammatory cytokines have an effect on dopamine release and proper function in the striatum [17]. Such findings support the notion that inflammatory cytokines lead to dopamine imbalance [14] in the dopaminergic pathways, namely the mesocorticolimbic and nigrostriatal systems. The dopamine imbalance hypothesis states that dopamine may play an important role in the perception of fatigue because it is central for cognition, motivation, and effortful behavior [1, 14]. In fact, individuals with high levels of fatigue have reduced mesocorticolimbic connectivity [16]. Dopamine is often referred to as the “reward neurotransmitter,” delicately balancing if a reward is worth the effort [1]. The effect of dopamine follows an inverted “u” shape, with optimal effect on the top of the curve; i.e., fatigue can be caused by too little as well as too much dopamine [14].
Predictors of post-aSAH fatigue
These hypotheses are relevant in relation to the presently identified predictors of fatigue. Our strongest predictor was use of nicotine. Nicotine addiction is created and maintained in the mesocorticolimbic system where it binds to nicotinic acetylcholine receptors and increases the firing rate and phasic bursts in midbrain dopamine neurons [2]. Chronic nicotine use leads to neuroadaptation and changes in dopamine homeostasis [2], possibly shifting dopamine levels to a point on the inverted “u” curve that promotes the development of fatigue. Nicotine is also a well-established risk factor for aSAH and clinical symptomatic vasospasm and delayed ischemic neurologic deficit in the course of aSAH [12]. Furthermore, nicotine aggravates the post-ischemic inflammatory response and thereby increases brain infarction size [5]. Common background factors for individuals with fatigue and nicotine users, like socio-economic factors and passive coping style, should also be considered.
Despite the large impact of nicotine on the same dopaminergic pathways that seem to be crucial in the development of fatigue, no other studies have investigated this correlation. To our knowledge, merely four previous studies [7, 26, 37, 41] have looked at the relationship between acute SAH-related factors and fatigue in the chronic phase, where three of the studies investigated their patients within 14 months after the hemorrhage. Rödholm et al. [41] found no significant association between fatigue (as defined by astheno-emotional disorder) and reaction level upon admission, amount of subarachnoid blood, or acute hydrocephalus. Passier et al. [37] found no significant association between fatigue assessed with the FSS and clinical status at admission, aneurysm localization, method of aneurysm repair, rebleed, secondary ischemia, or hydrocephalus. Khajeh et al. [26] found severity of SAH (in terms of World Federation of Neurosurgical Societies) to be associated with fatigue, but all other clinical characteristics (i.e., age, gender, body mass index, hydrocephalus, vasospasm, delayed cerebral ischemia (DCI), intraventricular hemorrhage (IVH), intraparenchymal hemorrhage, and rebleed) were not predictive of persistent fatigue. Buunk et al. [7] found a significant relationship between mental fatigue and external CSF drainage, but not with SAH type. Our results confirm in part their findings of lack of relation between clinical variables and fatigue; however, our univariable regression analysis identified GCS, rebleed, acute hydrocephalus, and large amounts of subarachnoid blood as relevant predictors of fatigue. This may be due to differences in evaluating the various variables or that our study cohort is more than three times larger than the patient populations examined in the earlier studies, rendering our results more robust.
Relevant predictors in our study, like LOCi, GCS, large amount of subarachnoid blood, and severe vasospasm, can also be linked to the inflammatory cytokine hypothesis with dopamine imbalance: when an aneurysm ruptures, there is an instant, vast increase in intracranial pressure (ICP), leading to cerebral circulatory arrest of varying length [35]. In some patients, the ICP increase is so brief or so moderate that they do not become unconscious, whereas up to half of aSAH patients experience some form of LOCi [20]. Prolonged LOCi often will lead to the patient being admitted with reduced GCS. If no LOCi occurred, or when awakening from LOCi, acute hydrocephalus can cause an additional and gradual decline in GCS. Presently, acute hydrocephalus was a relevant factor in univariable regression analysis, but did not prove to be an independent predictor like GCS, where we also saw that mean FSS increased the lower GCS was. Even very short-lasting circulatory arrest initiates a cascade of cellular pathophysiologic events as neuronal oxygen stores are depleted within 15–20 s [42]. Prolonged reduced consciousness in the setting of untreated acute aSAH is the result of local parenchymal damage and suboptimal cerebral perfusion pressure (CPP). This results in anaerobic glycolysis in the first 4–5 min and leads to depletion of brain glucose and adenosine triphosphate [33]. Such inflammation and downregulation of astrocytes leading to dysfunctional glutamate transmission have been attributed to mental fatigue after traumatic brain injury [24]. Microglia and endothelial cells become activated, and through complex mechanisms, proinflammatory cytokines (among others tumor necrosis factor-α, IL-6, and IL-8) are released [33].
The most feared complication during the first 2 weeks after aSAH is cerebral vasospasm [22]. Most patients experience some degree of vasospasm in conjunction with their aSAH, whereas usually only severe vasospasm becomes symptomatic due to reduced CPP. Without treatment adjustments to preserve adequate CPP, cerebral ischemia can occur or be aggravated [22]. Large amounts of subarachnoid blood represent a risk factor for developing vasospasm [22]. We found severe vasospasm to be an independent predictor of fatigue and large amount of subarachnoid blood was presently a predictor in the univariable analysis. Vasospasm after aSAH is not a purely mechanical event, but develops over days as an inflammatory response to blood degradation [15] and leads to a thickening of the arterial wall and thereby reduced arterial lumen. The inflammatory cytokines found in vasospasm comprise tumor necrosis factor-α, IL-1α, IL-1β, IL-6, and IL-8 [15], and in severe vasospasm, IL-6 seems to be predominant [22]. Hence, vasospasm is linked to the same inflammatory cytokines that are upregulated in patients with ischemia and/or reduced consciousness. Of these, IL-6 and tumor necrosis factor-α have also been directly related to the development of fatigue [40]. Inhibitors of tumor necrosis factor-α and antibodies against IL-1β have improved fatigue symptoms in patients with psoriasis, diabetes, and rheumatoid arthritis [25]. Substances ameliorating the dopamine imbalance, like methylphenidate or OSU6162, have also been mentioned as potential pharmacological therapies of fatigue [24]. Figure 4 illustrates the complex, interwoven relationship of the presently investigated variables of aSAH, which eventually may culminate in processes attributable to the development of fatigue.
Limitations
This study has several limitations. We used only one measure of fatigue, and there may have been other aspects of fatigue that remained under- or overestimated by the use of the FSS questionnaire. It is also uncertain whether administration of FSS by telephone interview in comparison with standard paper-and-pencil self-administration influenced the patients’ response style. We showed the frequency of fatigue to be remarkable stable up to 7 years after ictus, but time trajectories cannot be established with our cross-sectional study design. Although common to many stroke studies, generalizability of our findings was limited by excluding patients living at a nursing home, with pronounced cognitive sequelae or aphasia. This concerned only a small group of survivors (< 15%); however, it might have led to a selection bias where our estimate is an inaccurate reflection of fatigue prevalence in the overall aSAH population. On the other hand, we included patients from the entire range of aSAH severity. Finally, we assessed factors related to clinical status in the acute phase. Several other predictors such as premorbid personality traits (e.g., neuroticism, coping style), psychiatric comorbidity (e.g., mood disorder), and somatic comorbidity (e.g., sleep disorder, hypopituitarism, cancer, rheumatic diseases) were not measured. Future studies should include an even bigger range of possible factors related to fatigue. Altogether, these limitations should be considered when interpreting our findings. Still, our study included a large number of patients and is novel in that it could identify relevant clinical predictors of post-aSAH fatigue.
Conclusions
The prevalence of fatigue in the chronic phase after aSAH was 69.7%. Fatigue remained a common and stable symptom up to 7 years after the ictus. Nicotine use, reduced consciousness with GCS < 14 at admission, and severe vasospasm were independent predictors from the acute phase of aSAH that more than doubled the risk to develop post-aSAH fatigue. Inflammatory cytokines causing dopamine imbalance may be a common denominator for fatigue and the presently identified predictors.
Abbreviations
- ACA:
-
Anterior cerebral artery
- ACoA:
-
Anterior communicating artery
- aSAH:
-
Aneurysmal subarachnoid hemorrhage
- ATP:
-
Adenosine triphosphate
- CT:
-
Computed tomography
- CTA:
-
Computed tomography angiography
- CPP:
-
Cerebral perfusion pressure
- CSF:
-
Cerebrospinal fluid
- DCI:
-
Delayed cerebral ischemia
- FSS:
-
Fatigue Severity Scale
- GCS:
-
Glasgow Coma Scale
- HH:
-
Hunt and Hess Scale
- ICA:
-
Internal carotid artery
- ICH:
-
Intracerebral hemorrhage
- IVH:
-
Intraventricular hemorrhage
- ICP:
-
Intracranial pressure
- LOCi:
-
Loss of consciousness at ictus
- MCA:
-
Middle cerebral artery
- MRI:
-
Magnetic resonance imaging
- PTSD:
-
Post-traumatic stress disorder
- TCD:
-
Transcranial Doppler ultrasonography
References
Beeler JA, Mourra D (2018) To do or not to do: dopamine, affordability and the economics of opportunity:12. https://doi.org/10.3389/fnint.2018.00006
Biasi MD, Dani JA (2011) Reward, addiction, withdrawal to nicotine. 34:105–130. https://doi.org/10.1146/annurev-neuro-061010-113734
Boerboom W, Heijenbrok-Kal MH, Khajeh L, van Kooten F, Ribbers GM (2016) Long-term functioning of patients with aneurysmal subarachnoid hemorrhage: a 4-yr follow-up study. Am J Phy Med Rehabil 95:112–120. https://doi.org/10.1097/phm.0000000000000353
Bower JE, Ganz PA, Tao ML, Hu W, Belin TR, Sepah S, Cole S, Aziz N (2009) Inflammatory biomarkers and fatigue during radiation therapy for breast and prostate cancer. Clin Cancer Res 15:5534–5540. https://doi.org/10.1158/1078-0432.CCR-08-2584
Bradford ST, Stamatovic SM, Dondeti RS, Keep RF, Andjelkovic AV (2011) Nicotine aggravates the brain postischemic inflammatory response. Am J Physiol Heart Circ Physiol 300:H1518–H1529. https://doi.org/10.1152/ajpheart.00928.2010
Buunk AM, Groen RJM, Veenstra WS, Spikman JM (2015) Leisure and social participation in patients 4–10 years after aneurysmal subarachnoid haemorrhage. Brain Inj 29:1589–1596. https://doi.org/10.3109/02699052.2015.1073789
Buunk AM, Groen RJM, Wijbenga RA, Ziengs AL, Metzemaekers JDM, Dijk JMC, Spikman JM (2018) Mental versus physical fatigue after subarachnoid hemorrhage: differential associations with outcome. Eur J Neurol 25:1313–e1113. https://doi.org/10.1111/ene.13723
Chaudhuri A, Behan PO (2000) Fatigue and basal ganglia. J Neurol Sci 179:34–42. https://doi.org/10.1016/S0022-510X(00)00411-1
Chaudhuri A, Behan PO (2004) Fatigue in neurological disorders. Lancet 363:978–988. https://doi.org/10.1016/S0140-6736(04)15794-2
Cumming TB, Packer M, Kramer SF, English C (2016) The prevalence of fatigue after stroke: a systematic review and meta-analysis. 11:968–977. https://doi.org/10.1177/1747493016669861
De Doncker W, Dantzer R, Ormstad H, Kuppuswamy A (2018) Mechanisms of poststroke fatigue. J Neurol Neurosurg Psychiatry 89:287–293. https://doi.org/10.1136/jnnp-2017-316007
de Rooij NK, Rinkel GJ, Dankbaar JW, Frijns CJM (2013) Delayed cerebral ischemia after subarachnoid hemorrhage: a systematic review of clinical, laboratory, and radiological predictors. Stroke 44:43–54. https://doi.org/10.1161/STROKEAHA.112.674291
Dittner AJ, Wessely SC, Brown RG (2004) The assessment of fatigue: a practical guide for clinicians and researchers. J Psychosom Res 56:157–170. https://doi.org/10.1016/s0022-3999(03)00371-4
Dobryakova E, Genova HM, DeLuca J, Wylie GR (2015) The dopamine imbalance hypothesis of fatigue in multiple sclerosis and other neurological disorders. Front Neurol 6:52. https://doi.org/10.3389/fneur.2015.00052
Dumont AS, Dumont RJ, Chow MM, Lin C-l, Calisaneller T, Ley KF, Kassell NF, Lee KS (2003) Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery 53:123–135. https://doi.org/10.1227/01.NEU.0000068863.37133.9E
Engström M, Flensner G, Landtblom A-M, Ek A-C, Karlsson T (2013) Thalamo-striato-cortical determinants to fatigue in multiple sclerosis. Brain Behav 3:715–728. https://doi.org/10.1002/brb3.181
Felger JC, Miller AH (2012) Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol 33:315–327. https://doi.org/10.1016/j.yfrne.2012.09.003
Frontera JA, Claassen J, Schmidt JM, Wartenberg KE, Temes R, Connolly ES, Macdonald RL, Mayer SA (2006) Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 59:21–27. https://doi.org/10.1227/01.neu.0000243277.86222.6c
Hellawell DJ, Taylor R, Pentland B (1999) Persisting symptoms and carers’ views of outcome after subarachnoid haemorrhage. Clin Rehabil 13:333–340. https://doi.org/10.1191/026921599669500092
Hop JW, Rinkel GJ, Algra A, van Gijn J (1999) Initial loss of consciousness and risk of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Stroke 30:2268–2271. https://doi.org/10.1161/01.str.30.11.2268
Hunt WE, Hess RM (1968) Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 28:14–20. https://doi.org/10.3171/jns.1968.28.1.0014
Inagawa T (2016) Risk factors for cerebral vasospasm following aneurysmal subarachnoid hemorrhage: a review of the literature. World Neurosurg 85:56–76. https://doi.org/10.1016/j.wneu.2015.08.052
Jennett B, Bond M (1975) Assessment of outcome after severe brain damage: a practical scale. Lancet 305:480–484. https://doi.org/10.1016/S0140-6736(75)92830-5
Johansson B, Rönnbäck L (2014) Long-lasting mental fatigue after traumatic brain injury – a major problem most often neglected diagnostic criteria, assessment, relation to emotional and cognitive problems, cellular background, and aspects on treatment. In: Sadaka F (ed) Traumatic brain injury. INTECH, Rijeka. https://doi.org/10.5772/57311
Karshikoff B, Sundelin T, Lasselin J (2017) Role of inflammation in human fatigue: relevance of multidimensional assessments and potential neuronal mechanisms. 8. https://doi.org/10.3389/fimmu.2017.00021
Khajeh L, Ribbers GM, Heijenbrok-Kal MH, Blijdorp K, Dippel DW, Sneekes EM, van den Berg-Emons HJ, van der Lely AJ, Neggers SJ, van Kooten F (2016) The effect of hypopituitarism on fatigue after subarachnoid hemorrhage. Eur J Neurol 23:1269–1274. https://doi.org/10.1111/ene.13014
Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD (1989) The fatigue severity scale: application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol 46:1121–1123. https://doi.org/10.1001/archneur.1989.00520460115022
Kutlubaev MA, Barugh AJ, Mead GE (2012) Fatigue after subarachnoid haemorrhage: a systematic review. J Psychosom Res 72:305–310. https://doi.org/10.1016/j.jpsychores.2011.12.008
Le Roux PD, Winn HR (1999) Intracranial aneurysms and subarachnoid hemorrhage management of the poor grade patient. In: Acta Neurochir Suppl, vol 72. 1999/05/25 edn., pp 7-26
Lerdal A, Wahl AK, Rustoen T, Hanestad BR, Moum T (2005) Fatigue in the general population: a translation and test of the psychometric properties of the Norwegian version of the fatigue severity scale. Scand J Public Health 33:123–130. https://doi.org/10.1080/14034940410028406
Lindegaard KF, Nornes H, Bakke SJ, Sorteberg W, Nakstad P (1988) Cerebral vasospasm after subarachnoid haemorrhage investigated by means of transcranial Doppler ultrasound. Acta Neurochir Suppl 42:81–84. https://doi.org/10.1007/978-3-7091-8975-7_16
Mead G, Lynch J, Greig C, Young A, Lewis S, Sharpe M (2007) Evaluation of fatigue scales in stroke patients. Stroke 38:2090–2095. https://doi.org/10.1161/STROKEAHA.106.478941
Michenfelder J, Theye RJ (1970) The effects of anesthesia and hypothermia on canine cerebral ATP and lactate during anoxia produced by decapitation. Anesthesiology 33:430–439. https://doi.org/10.1097/00000542-197010000-00013
Nordenmark TH, Karic T, Røe C, Sorteberg W, Sorteberg A (2019) The post-aSAH syndrome: a self-reported cluster of symptoms in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg:1–10. https://doi.org/10.3171/2019.1.JNS183168
Nornes H (1973) The role of intracranial pressure in the arrest of hemorrhage in patients with ruptured intracranial aneurysm. 39:226. https://doi.org/10.3171/jns.1973.39.2.0226
Passier PE, Visser-Meily JM, van Zandvoort MJ, Post MW, Rinkel GJ, van Heugten C (2010) Prevalence and determinants of cognitive complaints after aneurysmal subarachnoid hemorrhage. Cerebrovasc Dis (Basel, Switzerland) 29:557–563. https://doi.org/10.1159/000306642
Passier PE, Post MWM, van Zandvoort MJE, Rinkel GJE, Lindeman E, Visser-Meily JMA (2011) Predicting fatigue 1 year after aneurysmal subarachnoid hemorrhage. J Neurol 258:1091–1097. https://doi.org/10.1007/s00415-010-5891-y
Passier PE, Visser-Meily JM, Rinkel GJ, Lindeman E, Post MW (2011) Life satisfaction and return to work after aneurysmal subarachnoid hemorrhage. J Stroke Cerebrovasc Dis 20:324–329. https://doi.org/10.1016/j.jstrokecerebrovasdis.2010.02.001
Powell J, Kitchen N, Heslin J, Greenwood R (2004) Psychosocial outcomes at 18 months after good neurological recovery from aneurysmal subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry 75:1119–1124. https://doi.org/10.1136/jnnp.2002.000414
Raison CL, Lin J-MS, Reeves WC (2009) Association of peripheral inflammatory markers with chronic fatigue in a population-based sample. Brain Behav Immun 23:327–337. https://doi.org/10.1016/j.bbi.2008.11.005
Rödholm M, Starmark JE, Ekholm S, Von Essen C (2002) Organic psychiatric disorders after aneurysmal SAH: outcome and associations with age, bleeding severity, and arterial hypertension. Acta Neurol Scand 106:8–18. https://doi.org/10.1034/j.1600-0404.2002.01128.x
Sanganalmath SK, Gopal P, Parker JR, Downs RK, Parker JC, Dawn B (2017) Global cerebral ischemia due to circulatory arrest: insights into cellular pathophysiology and diagnostic modalities. Mol Cell Biochem 426:111–127. https://doi.org/10.1007/s11010-016-2885-9
Vetkas A, Lepik T, Eilat T, Rätsep T, Asser T (2013) Emotional health and quality of life after aneurysmal subarachnoid hemorrhage. Acta Neurochir 155:1107–1114. https://doi.org/10.1007/s00701-013-1683-3
Visser-Meily JM, Rhebergen ML, Rinkel GJ, van Zandvoort MJ, Post MW (2009) Long-term health-related quality of life after aneurysmal subarachnoid hemorrhage: relationship with psychological symptoms and personality characteristics. Stroke 40:1526–1529. https://doi.org/10.1161/strokeaha.108.531277
Acknowledgments
We are grateful to the patients who participated.
Funding
Open Access funding provided by University of Oslo (incl Oslo University Hospital). EW received funding from SveaNor Fastigheter AB, Haugans Hus Invest AS, and Hathon Holding AS. None of the funding sources had any role in the study design, data analysis, data interpretation, the preparation of the manuscript, or the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Comments
Let us say what is intuitive; in aSAH and all serious neurosurgical diseases that we treat. Sicker patients with more comorbidities have more protracted recovery periods. This is known to all experienced neurosurgeons and is part of our daily lives.
Now here the authors have applied a scientific rationale in a prospective study to further characterize our intuitive observations. The scientific aim was to study many independent factors to see which could contribute to long-term post SAH fatigue on a standard measure. In their paper, univariable analysis identified current nicotine use, loss of consciousness at ictus, rebleed prior to aneurysm repair, reduced consciousness to Glasgow Coma Scale (GCS) < 14, large amounts of subarachnoid blood, the presence of acute hydrocephalus, and severe vasospasm as factors that were significantly associated with fatigue. In multivariable analysis, current nicotine use, reduced GCS, and severe vasospasm were independent predictors that all more than doubled the risk to develop post-aSAH fatigue.
Experienced surgeons would have predicted this. None of these are really variables that we can control. Nonetheless, it is useful to have this data scientifically quantified in a proper study, if only to give us a better foundation for discussions of expectations with patients and family.
Christopher Miranda Loftus
PA, USA
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Vascular Neurosurgery - Aneurysm
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Western, E., Sorteberg, A., Brunborg, C. et al. Prevalence and predictors of fatigue after aneurysmal subarachnoid hemorrhage. Acta Neurochir 162, 3107–3116 (2020). https://doi.org/10.1007/s00701-020-04538-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-020-04538-9