
Abstract
The cashew, Anacardium occidentale, is a globally important tropical fruit tree, but little is known about its natural infraspecific systematics. Wild Brazilian populations occur in the cerrado biome and coastal restinga vegetation. We investigated whether wild coastal and domesticated populations could be distinguished genetically using inter-simple repeat molecular markers (ISSRs). In total, 94 polymorphic loci from five primers were used to characterise genetic diversity, structure and differentiation in four wild restinga populations and four domesticated ones from eight localities in Piauí state (30 individuals per population). Genetic diversity was greater overall in wild (%P: 57.2%, I: 0.24, He : 0.15) than domesticated populations (%P: 49.5%, I: 0.19, He : 0.12). Significant structure was observed among the eight populations (between-population variance 22%, ΦPT = 0.217, P ≥ 0.001), but only weak distinctions between wild and domesticated groups. Cluster and principal coordinate analyses showed marked genetic disparity in populations. No correlation of genetic and geographical inter-population distance was found (Mantel test, r = 0.02032, P = 0.4436). Bayesian analysis found an eight-group optimal model (ΔK = 50.2, K = 8), which mostly corresponded to sampled populations. Wild populations show strong genetic heterogeneity within a small geographical area despite probable gene flow between them. Within-population genetic diversity of wild plants varied considerably and was lower where extractive activities by local people are most intense (Labino population). The study underlines the importance of wild populations as in situ genetic reserves and the urgent need for further studies to support their conservation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The cashew or caju (Anacardium occidentale L.) is a fruit tree cultivated throughout the tropics but native to South America (Johnson 1973; Mitchell and Mori 1987). It has a significant agronomic role globally, especially for the edible seed. The hypocarp or pseudofruit is eaten fresh or used to manufacture sweets or pulp for juices and other drinks, and the residue from processing is used as a component of animal feed. The nut shell is the source of cashew nut shell liquid (CNSL), valuable in the chemical industry for the manufacture of dyes, lubricants and cosmetics. Tannins—used widely in industrial applications—are extracted from branches, leaves, the testa of the kernel (seed) and the hypocarp residue (USAID-BRASIL 2006).
Anacardium occidentale was first introduced from Brazil into India and Africa (Nigeria) by the Portuguese from the sixteenth century to early seventeenth century where it spread spontaneously as well as through human agency, forming both wild and domesticated populations (Johnson 1973; Archak et al. 2009; Aliyu 2012; Adeigbe et al. 2015). Its growing economic importance in the twentieth century resulted in the establishment of national and regional germplasm collections that conserve genetic diversity and provide material for breeding (Aliyu 2012; Mohana et al. 2018). These germplasm collections have been the basis of many studies that aimed to compare the genetic diversity of cashew accessions from different regions. Some used morphological markers (see references in Andrade et al. 2019; Bionoset 2019), but genotyping with molecular markers such as isozymes, RAPDs, ISSRs, AFLP, microsatellites and ITS sequence data is now more usual, sometimes in combination with morphological descriptors; these studies have been carried out predominantly on material from India (e.g. Dhanaraj et al. 2002; Rout et al. 2002; Samal et al. 2002; Archak et al. 2003a, b; Samal et al. 2003; Desai 2008; Archak et al. 2009; Thimmappaiah et al. 2009; Dashmohapatra et al. 2014; Sethi 2015; Jena et al. 2016), Brazil (e.g. Barros 1991; Cavalcanti and Wilkinson 2007; Pessoni 2007; Amaral et al. 2017; Borges et al. 2018), Nigeria (e.g. Aliyu and Awopetu 2007) and Tanzania (e.g. Mneney et al. 2001; Croxford et al. 2006).
Studies have shown that in Asia and Africa cashews have a relatively narrow genetic base (Aliyu 2012; Archak et al. 2009) and diversity within the natural South American range of the species would be expected to be greatest. However, the genetic and phenotypic variability of natural populations in Brazil remains poorly known as there are few such studies (Andrade et al. 2019). Biosystematic investigations of A. occidentale have been hampered by difficulties in determining its species limits and the status of populations as natural, naturalised or domesticated (Johnson 1972, 1973; Mitchell and Mori 1987). As regards the former, various publications have reported on plants determined as A. microcarpum Ducke or A. othonianum Rizzini (see Andrade et al. 2019 for further details), which taxonomists consider conspecific with A. occidentale (Mitchell and Mori 1987; Luz et al. 2019). In regard to infraspecific taxonomy, Mitchell and Mori (1987) discussed the long history of human use of A. occidentale and its transport around the globe and put forward an informal classification of this species consisting of two natural forms centred in Brazil, a cerrado ecotype occurring in the interior and a restinga ecotype along the coast, both known locally as “cajuí”. The coastal populations can be regarded as wild wherever they are a component of natural restinga vegetation on sandy substrates (Araujo et al. 2019), including stabilised dune fields (Johnson 1972, 1973; Lima 1986; Mitchell and Mori 1987; Barros 1991; Freitas and Paxton 1998; Rufino et al. 2007; Andrade et al. 2019).
The present study of A. occidentale is an investigation of genetic diversity that focusses primarily on restinga ecotype populations occurring in coastal Piauí state. It is one of the first to explore genetic data using statistically adequate samples made directly from natural populations. We sought in the first place to establish whether there was a clear difference between wild and domesticated plants at the population level and secondly to generate diversity data to characterise the wild populations genetically and compare the patterns found to those from domesticated plants. It complements morphometric studies by Vieira et al. (2014) and Andrade et al. (2019) and includes some of the same populations. We used ISSR molecular markers (inter-simple sequence repeat), widely deployed in studies of economically important species and their wild relatives that focus on genotype identification, genetic conservation and cultivar development (e.g. Nunes et al. 2013; Martins et al. 2014; Oliveira et al. 2014; Rodrigues et al. 2015; Silva et al. 2014; Carmo et al. 2017; Wu et al. 2019).
Natural restinga cashew populations appear to play a key ecological role in the establishment and maintenance of the woody restinga vegetation that develops over dune fields along the coast of northeast Brazil (Fernandes et al. 1996; Santos-Filho et al. 2010). These abundant wild populations are subject to extractive collection of their fruits, an important seasonal source of income for local people (Rufino 2004; Rufino et al. 2007, 2008; Crespo and Souza 2014), but the effect of this activity on population genetic diversity has not been studied. The coastal habitats are under increasing pressure from agricultural, industrial and urban development, highlighting the need for in situ conservation strategies. This study arose as a response to these considerations. Its aim is to contribute information useful for the future management and in situ conservation of remaining natural populations. Although part of a genetic resource of global importance, these wild plant communities urgently require further management and protection.
Materials and methods
Populations and sampling
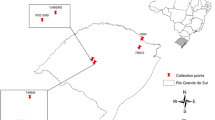
Samples were collected from eight populations in different localities in northern Piauí state in Brazil (Table 1, Fig. 1). The populations from the localities Cajueiro da Praia (CP), Cocal (CL), Luzilândia (LU) and Rosápolis (RO) were domesticated genotypes of A. occidentale identified by local people as “caju”. Those from Cal (CA), Labino (LA), Pedra do Sal (PS) and Tatus (TU) were natural populations on stabilised dunes identified locally as “cajuí” and consisted of wild genotypes of the restinga ecotype of A. occidentale. Collections were made from August to October 2015 during the main flowering and fruiting season. Balanced and statistically robust population sampling was prioritised. Young leaves were gathered from 30 different plants in each population and stored in silica gel, making a total sample of 240 individuals across the eight populations studied. Individuals more than 10 m apart were selected for sampling because plants in natural restinga populations are usually mixed together with other woody species in thickets of various sizes and degrees of isolation.

Map showing geographical location of the sampled populations of Anacardium occidentale. Northern coast of Brazil showing location of populations in Piauí state; wild populations shown as red dots: CA Cal, LA Labino, PS Pedra do Sal, TU Tatus; domesticated populations shown as white dots: CP Cajueiro da Praia, CL Cocal, LU Luzilândia, RO Rosápolis. Inset shows Brazil and the study location in South America, viewed from space at altitude 10,391 km. All base images from Google Earth (Google Earth 2019; downloaded 31 March 2019)
DNA extraction
Genomic DNA was extracted using the method described by Doyle and Doyle (1990) with some modifications to obtain optimal DNA quality. Approximately 20 mg of young leaves were macerated with extraction buffer in the proportion of 800 µL of CTAB 2× and 4 µL β-mercaptoethanol [CTAB 2%, Tris–HCL 0.1 mM (pH 8.0), EDTA 20 mM (pH 8.0), NaCl 1.4 M and β-mercaptoethanol 2%] previously heated in a water bath at 60 °C for 10 min. Extraction buffer was then added and the mixture heated for 20 min at 60 °C. After cooling, 800 µL of a 24:1 solution of chloroform and isoamyl alcohol were added to the samples, homogenised in a shaker for 1 h and centrifuged for 10 min at 13,000 rpm. Part of the resulting supernatant (~ 400 µL) was transferred to a new tube, to which was added two-thirds of its volume of isopropanol (~ 300 µL), and then carefully mixed by inversion and stored overnight in a freezer. The samples were then centrifuged at 13,000 rpm for 5 min, which brought about precipitation of the DNA. 1000 µL of 70% ethanol was added to the pellet, followed by centrifugation for 5 min at 13,000 rpm; these two operations were repeated three times. The DNA obtained was resuspended in 100 µL of TE solution [Tris–HCl 10 mM (pH 8.0) and EDTA 0.1 mM] for 24 h on the laboratory bench, or until the pellet had blended into the solution.
The DNA samples were quantified using a BioSpec-nano (Kyoto, Japan) spectrophotometer and then diluted to a concentration of 25 ng/μL. To confirm their quality, some samples were quantified using the method of visualisation in bands by agarose gel electrophoresis at a concentration of 1%, prepared with TBE 1× buffer (Tris-Borato-EDTA) and stained with GelRed (Biotium®, California, USA) at 1×. Lambda (λ) phage DNA at a concentration of 100 ng/μL was used for comparison.
Polymerase chain reaction (PCR) and ISSR primer selection
The PCR reaction was carried out with the TopTaq Master Mix kit (Qiagen, Maryland, USA). The mix was prepared with a total volume of 10 μL according to the following proportions: 4 μL of TopTaq polymerase, 4.7 μL of H2O-free RNase, 0.8 μL of CoralLoad, and 0.5 μL of primer. For the PCR reaction 9 μL of the mix and 1 μL of genomic DNA (25 ng/μL) were used. The amplification reactions were carried out in a Tprofessional Thermocycler (Biometra®, Göttingen, Germany) with 96-sample capacity using the following parameters: an initial denaturing at 94 °C for 1.5 min, followed by 35 denaturing cycles at 94 °C for 40 s, annealing for 45 s at the required temperature for the primer being used, an extension at 72 °C for 1.5 min and a final stage of extension at 72 °C for 10 min. The PCR products were then run by electrophoresis on a 1.5% agarose gel in TBE buffer (Tris-Borato-EDTA) 1×, at a constant current of 100 V. For the electrophoresis runs, 10 μL of PCR product was used with 3 μL of BLUE and 2 μL of GelRed (Biotium®, California, USA) at 1×. The same quantities were used for the control group. In all the gels, a marker with known molecular weight was added for comparison: 5 μL of Ladder 100 pb (Invitrogen, California, US) was added into the channel of each gel. The gels were then visualised in a UV transilluminator (Loccus Biotecnologia, São Paulo, Brazil) and photo-documented.
Tests were carried out with 18 ISSR primers of 14–20 nucleotides (UBC 807, UBC 810, UBC 811, UBC 813, UBC 814, UBC 824, UBC 825, UBC 843, UBC 844, UBC 847, UBC 853, UBC 860, UBC 899, BECKY, MANY, MAO, OMAR, TERRY) length in order to optimise and select the primers with the best pattern of amplification (Online Resource 1). Two individuals from each collecting locality were used for these tests to verify the existence of polymorphism. After establishing which primers had the best amplification, the technique was applied to all 30 individuals of each population. Tests for reproducibility were carried out by repeating the laboratory processes for two of the five primers in three replications from six individuals drawn at random from five of the eight populations. The PCR products were run on separate gels and the markers were scored separately. Overall genotyping error was computed by using the mismatch error rate formula (Vašek et al. 2017) for all 270 comparisons of paired replicate binary vectors.
Data analysis
The fragments produced by the genomic DNA amplification of each sample were used as the data for this study. The genotyping of each individual was carried out by direct inspection of the fragments that are represented in the gel images as bands (Online Resource 2). Only unequivocally distinct fragments with higher intensity were recorded, while those with low intensity or poor definition were not included. Each recorded fragment was designated as a single unique character and coded as “1” when present and “0” when absent. The resulting binary matrix was used in the statistical analyses (Online Resource 3).
The percentage polymorphism of each primer was obtained as the ratio between the number of polymorphic bands and the total number of bands. The software GenAlEx 6.502 (Peakall and Smouse 2012a) was used to compute the percentage polymorphism per population (%P), obtained by dividing the number of polymorphic bands in each population by the total number of bands. Other parameters of genetic diversity calculated were Shannon’s index (I) and expected heterozygosity (He) based on Nei (1978).
The estimation of the proportions of within- and between-population genetic variability was made using analysis of molecular variance (AMOVA: Excoffier et al. 1992), as implemented in GenAlEx 6.502 (Peakall and Smouse 2012a). In this software, the calculation is based on the parameter ΦPT (an analogue of FST), which is more appropriate for carrying out AMOVA using dominant markers (Peakall and Smouse 2012b, 2015). Multiple comparisons of the ΦPT values were calculated in GenAlEx for all population pairs using a permutation test (999 replications) to compute their P values.
Genetic divergence between populations was investigated using the unbiased genetic distance and identity measures of Nei (1978). These calculations were carried out using GenAlEx 6.502 (Peakall and Smouse 2012a). The software PAST 2.17c (Hammer et al. 2001) was used to construct a UPGMA (unweighted pair group method with arithmetic mean) dendrogram based on between-population Nei’s genetic distance (Nei 1978), and to compute the correlation between inter-population genetic (both Nei’s distance and ΦPT values) and geographical distances (metres) using the Mantel test (9999 permutations).
Different views of inter-population similarities were obtained using principal coordinate analysis (PCoA). Genetic distance matrices were computed using GenAlEx version 6.502 (Peakall and Smouse 2012a). For the analysis of all individuals, a matrix of inter-individual genetic distances (GD) was used, as defined for binary data by Peakall and Smouse (2015). Inter-population genetic distances were computed using between-population Nei’s genetic distances. Ordinations and minimum spanning trees were computed in PAST version 2.17c (Hammer et al. 2001).
Bayesian analysis was used to investigate genetic structure with the software STRUCTURE (Pritchard et al. 2000). To determine the optimal number of genetic clusters (K), ten simulation runs were computed for each value of K from 1 to 20. The admixture model was used for this analysis since it assumes that each individual has mixed ancestry (Pritchard et al. 2010), a likely scenario in this highly outcrossing species. The allelic frequencies were estimated by 500,000 MCMC (Markov Chain Monte Carlo) replications after a burn-in of 50,000 replications. The procedure described by Evanno et al. (2005) was used to determine the optimal number (K) of genetic clusters, as implemented in the software STRUCTURE HARVESTER v. 0.6.9 (Earl and Vonholdt 2012). This is the K number corresponding to the modal value of delta K (ΔK), a parameter which, for each K, is the mean of the absolute values of the second-order rate of change of the likelihood function L(K) divided by the standard deviation of L(K) (Evanno et al. 2005). ΔK is thus a measure of the greatest change in the value of the mean likelihood of the data across a range of values of K, corrected for the variance obtained among the replicate runs for each K value.
Results
Primer polymorphism
Of the 18 primers tested, five (UBC 813, UBC 825, UBC 847, UBC 860 and Many) were selected and used in this study. These primers had the best pattern of amplification as regards polymorphism, quality and resolution of the bands (Table 2). The five primers generated a total of 94 bands (loci), varying in length from 200 to 2000 bp. All the primers used exhibited 100% polymorphism. The primer with the least number of polymorphic loci was UBC 825 (17) and those with the greatest were UBC 847 and Many, with 20 loci each (Table 2). The result of the reproducibility tests was an overall genotyping error of 6.7% (18 mismatches from 270 duplicate comparisons).
Genetic diversity within populations
The percentages of polymorphic loci (%P) found in each population varied (Table 1, Fig. 1), the highest were in TU and PS with 63.83% and 67.02%, respectively, and the lowest in the populations CP, LA and LU with 40.43%, 45.74% and 46.87%, respectively. The populations at CL and CA were the same at 52.13%, and RO showed somewhat higher polymorphism at 58.51%. These values are consistent with those obtained with the genetic diversity estimators Shannon’s index (I) and expected heterozygosity (He), which were greatest in the TU and PS populations with values, respectively, of 0.261 (I), 0.164 (He) and 0.285 (I), 0.180 (He). The populations at CP, LA and LU had the lowest values: 0.166 (I), 0.104 (He); 0.170 (I), 0.104 (He); 0.172 (I), 0.103 (He), respectively, and the populations CA, RO and CL showed intermediate values of 0.227 (I), 0.145 (He); 0.216 (I), 0.132 (He); 0.215 (I), 0.137 (He), respectively.
The results indicated that the three wild populations at PS, TU and CA have the greatest within-population genetic diversity, while that at LA is similar to the less diverse populations of domesticated cashew at CP and LU. The wild populations (CA, LA, PS, TU) have a wider range of diversity than domesticated ones (CL, CP, LU, RO; Table 1). The mean values of the parameters of genetic diversity are lower in the four domesticated populations when treated as a single group (%P: 85.11%, I: 0.227, He: 0.132) than in the four wild populations similarly treated (%P: 89.36%, I: 0.274, He: 0.161).
Genetic differentiation between populations
The results of the analysis of molecular variance (AMOVA, Table 3) showed that genetic variability was greater within populations (78%) than between them (22%). The value of the ΦPT fixation index (ΦPT = 0.217, P ≥ 0.001) showed that there are significant between-population differences (Table 3) and in the multiple comparisons of ΦPT values, all population pairs were found to be significantly different (P ≥ 0.001, Online Resource 5).
Nei’s genetic distance (Nei 1978), a measure of genetic divergence among populations, varied from 0.006 to 0.067, with the lowest values observed between CP and LU and the highest between CA and CL (Online Resource 6). The UPGMA dendrogram based on this distance showed that the CL and CA populations were well differentiated from the others (Online Resource 4, 6). The PS population was less so, and LU and CP formed a well-separated pair. The remaining populations RO, LA and TU were rather similar to one another. The composition of these subgroups in the dendrogram suggests little relationship between inter-population geographical and genetic distances, and this was corroborated by the non-significant result of the Mantel test (with Nei’s genetic distance r = 0.02032, P = 0.4436; with ΦPT values r = −0.02032, P = 0.4674, Online Resource 7). The population at LU (Online Resource 4) was genetically most similar to the most distant population CP (133 km) and genetically most distant from CL, the geographically closest population (89 km, Fig. 1).
The principal coordinate analysis (PCoA) of population centroids also used Nei’s distance. In these ordinations (Fig. 2), the first three axes express 87.8% of the total variance of the data set and thus can be taken to show the most important patterns. They show that the most divergent populations are CA, PS and CL, the first two being wild populations of the restinga ecotype and CL belonging to the domesticated genotypes, supporting the inference of greater genetic diversity in wild populations. The minimum spanning tree (MST) links the points to their closest neighbours in the Nei’s distance matrix and thus compensates for the more distorted view of relationships inevitable in a two-dimensional ordination. The MST links CA to TU and PS to RO.

Principal coordinate analysis (PCoA) using Nei’s (1978) distance of eight populations of wild and domesticated Anacardium occidentale, computed using Genalex 6.502 (Peakall and Smouse 2012a) and PAST version 2.17c (Hammer et al. 2001). a Ordination on principal coordinates 1 (56.94% variance) and 2 (19.26% variance). b Ordination on principal coordinates 1 and 3 (11.6% variance). The lines represent the minimum spanning tree. Population codes as in Fig. 1. Wild populations: red and bold font; domesticated populations: black and normal font
The PCoA of all the individuals of the populations (Fig. 3) using genetic distance for binary data (GD) showed considerable overlap between populations, but on coordinates 1 and 2, the populations CL, PS, CA and TU were partially separated from a denser group consisting of the superimposed populations at RO, LU, CP and LA.

Principal coordinate analysis (PCoA) using the genetic distance GD between all individuals of eight populations of wild and domesticated Anacardium occidentale, computed using Genalex 6.502 (Peakall and Smouse 2012a). a Ordination on principal coordinates 1 (8% variance) and 2 (7.1% variance). b Ordination on principal coordinates 1 and 3 (6.1% variance). Wild populations: Cal, Labino, Pedra do Sal, Tatus. Domesticated populations: Cajueiro da Praia, Cocal, Luzilândia, Rosápolis
In the Bayesian simulation analysis carried out with STRUCTURE software, the optimal number of genetic groups (K) was found to be eight (Fig. 4). The bar diagram of the eight-cluster model (Fig. 4) showed that six of the genetic groups corresponded to the populations, one (orange–brown) was predominantly common to the LU and CP populations and one (dark blue) was scattered throughout the populations. The genetic similarity between LU and CP was consistent with the result given by hierarchical cluster analysis (Online Resource 4). Examination of the bar plots of other models analysed (K = 3–20) showed that the CL population was consistently distinct from all the rest and showed very little mixture. The scattered (dark blue) genetic pattern was also least present in the CL population and most conspicuous in RO, LU, CP and LA.

Bayesian genetic structure analysis. a Optimal number of genetic clusters (K = 8) in eight populations of wild and domesticated Anacardium occidentale using ΔK optimality criterion; computed with STRUCTURE HARVESTER (Earl and Vonholdt 2012). b Optimal genetic structure (K = 8 genetic clusters) of the eight populations obtained by Bayesian analysis. Each genetic cluster represented by a different colour. Black rectangle borders mark population boundaries: wild and domesticated populations designated as in legend of Fig. 3. Each population rectangle is composed of narrow vertical bars representing individuals. The length of each colour in an individual bar as measured on the y-axis is proportional to its probability of membership in the genetic cluster indicated by that colour. Computed with STRUCTURE software (Pritchard et al. 2000)
Discussion
Wild populations in the same region sampled by Andrade et al. (2019) in a morphometric study could be differentiated statistically as a category from domesticated ones, and their similarity was significantly correlated with the geographical distance between them. In contrast, no such distinction between wild and domesticated populations was observed using ISSR molecular marker data nor any correlation with geography. These results suggest that morphological similarity may not be a reliable guide to genetic diversity within this species. The genetic data also reveal much greater disparity between the wild populations than domesticated ones (Fig. 2), and at the same time, the overall within-population diversity was greater in wild populations (Table 1). The population growing at Labino contradicted this pattern, having much lower diversity. This may be caused by the more intense extractive fruit collection at this locality (Rufino et al. 2008) and possible genetic erosion comparable to that reported by Cota et al. (2017) in wild populations of A. humile A.St.-Hil.
ISSRs are regarded as less reproducible than AFLPs by various authors (e.g. Crawford et al. 2012), but others argue that this is offset by their cost-effectiveness and simpler technical implementation (Ng and Tan 2015). These markers continue to be used especially in genetic structure studies in economically important plant species (e.g. Kumar and Agrawal 2017; Wu et al. 2019). Most studies of genotyping error in dominant markers have been carried out on AFLP data. Vašek et al. (2017), in a recent study, found that error rate affected descriptive parameters of diversity such as He, %P and ΦPT more strongly than the results of Bayesian STRUCTURE analysis. We therefore judge that our observed genotyping error (6.7%, which compares to the 5% maximum AFLP error rate used by Vašek et al. 2017) is unlikely to have affected the optimal 8-group genetic structure or the relative values of the genetic diversity parameters among the wild and domesticated populations. However, comparison of these values with those of other studies of this and other species should only be made with caution.
The distinctness of most of the populations as genetic groups was confirmed both by Bayesian analysis and AMOVA, differing from the ISSR studies of Borges (2015), Gomes (2017) and Borges et al. (2018) which found less genetic differentiation between populations. However, the Bayesian analysis also provided evidence of inter-population gene flow, which is to be expected in a species which is regarded as highly outcrossing. Bees are reported as the main pollination vectors (Mitchell and Mori 1987; Paulino 1992; Freitas and Paxton 1998; Bhattacharya 2004; Ribeiro et al. 2008) and fruit-feeding bats as dispersers (Mitchell and Mori 1987). Mitchell and Mori (1987) also suggested that inter-species crossing between sympatric A. occidentale, A. humile and A. nanum A.St.-Hil. may occur in the central Brazilian Cerrado because of lack of intrinsic barriers. Human dispersal must affect genetic patterns in domesticated populations; transport of genotypes by local farmers might explain the similarity between the Luzilândia (LU) and Cajueiro da Praia (CP) populations, the latter being a locality well known for its giant cashew tree (Amaral et al. 2017).
The genetic studies of Borges (2015), Cota et al. (2017), Gomes (2017) and Borges et al. (2018) all agree with ours in the absence of correlation between geographical and genetic distance, suggesting lack of an isolation-by-distance effect. These studies also support the view that significant inter-population gene flow occurs, including between wild and domesticated ones. The study of A. humile by Cota et al. (2017), based on co-dominant microsatellite markers, suggested another source of genetic structure. They observed significant inbreeding within most populations, and this led them to propose that the natural clumping of the plants of this species would promote crossing between genetically very similar flowers and the consequently increased inbreeding levels would lead to a stronger spatial genetic structure of the populations. Clumped physiognomy is also a characteristic of populations of the restinga ecotype of A. occidentale (Andrade et al. 2019), and the inbreeding effect reported by Cota et al. (2017) is therefore a possible contribution to their genetic structure which should be investigated in the future.
Our study adds to current knowledge of the genetic diversity and structure of wild cashew populations in northeast Brazil, but a robust understanding of the genetic patterns is still a future goal. Basic taxonomic information such as the distinction between cerrado and restinga ecotypes (Mitchell and Mori 1987, Andrade et al. 2019) and an accurate estimate of the geographical range of wild cashews remain to be fully established. Our study, like those of Cota et al. (2017) and Gomes (2017), indicates that intra-specific geographical patterns are complex. Wild populations of A. occidentale showed high genetic diversity within a small area and most were genetically distinct, but no consistent geographical genetic pattern has yet emerged. Along the northern coast of northeast Brazil, wild cashew populations are present in large areas of restinga habitat of the states of Maranhão, Piauí, Ceará and Rio Grande do Norte, and represent a major and as yet poorly investigated resource for researchers working on genetic diversity of cashews. Some of these areas are relatively remote and likely to be less influenced by gene flow from domesticated orchards, increasing their potential for scientific investigation.
These considerations highlight the need for in situ conservation of wild cashews. Although ex situ germplasm collections are crucial for genetic diversity conservation in A. occidentale, they can provide only a simplified overview of the genetic basis of the species. In situ conservation is an important complement to germplasm collections if the widest possible genetic basis for the future agronomic development is to be ensured (Kell et al. 2012; Whitlock et al. 2016). In order to make the best choice of areas to conserve, it is clearly necessary to carry out more extensive genetic surveys at population level. In Brazil, in situ conservation has other benefits as well. Not only could wild cashews provide greater long-term economic benefit for local people if based on sound knowledge of genetic diversity, but A. occidentale is a keystone woody species (Santos-Filho et al. 2010) of the restinga vegetation which secures large areas of underlying ancient sand deposits (Guedes et al. 2017). Active dunes cause serious problems for dwellings and businesses in this region, and the effect of removing natural vegetation on reactivation of dune systems is an issue that also needs further research and is likely to become more important with increasing real estate and industrial development along the coast.
Conclusions
We conclude that the natural populations of the restinga ecotype of A. occidentale in the coastal regions of northeast Brazil represent a genetic resource of great importance for local people and for the future of the cashew agronomic industry. More extensive surveys of these populations are required, and future studies should include analysis of co-dominant markers, since the spatial genetic structure of the populations can only be fully understood if inbreeding can be estimated with confidence. This will provide information needed to formulate more accurately tuned in situ and ex situ conservation measures in restinga areas, reinforce management policy for existing and future conservation units and target the further selection of wild genotype accessions for Brazil’s cashew germplasm banks.
Data availability
The data set generated and analysed in the present study is presented in Online Resource 3.
References
Adeigbe OO, Olasupo FO, Adewale BD, Muyiwa AA (2015) A review on cashew research and production in Nigeria in the last four decades. Sci Res Essays 10:196–209. https://doi.org/10.5897/SRE2014.5953
Aliyu OM (2012) Genetic diversity of Nigerian cashew germplasm. In: Caliskan M (ed) Genetic diversity in plants. Intech, Rijeka, pp 163–184
Aliyu OM, Awopetu JA (2007) Assessment of genetic diversity in three populations of cashew (Anacardium occidentale L.) using protein-isoenzyme-electrophoretic analysis. Genet Resources Crop Evol 54:1489–1497. https://doi.org/10.1007/s10722-006-9138-9
Amaral FPM, Sá GH, Filgueiras LA, Santos Filho FS, Santos Soares CJR, Amaral MPM, Valente SES, Mendes AN (2017) Genetics analysis of the biggest cashew tree in the world. Genet Molec Res 16:1–7, gmr16039817
Andrade IM, Nascimento JDAO, Sousa MV, Santos JO, Mayo SJ (2019) A morphometric study of the restinga ecotype of Anacardium occidentale (Anacardiaceae): wild coastal cashew populations from Piauí, Northeast Brazil. Feddes Repert 130:89–116. https://doi.org/10.1002/fedr.201800024
Araujo DSD, Sá CFC, Fonseca-Kruel VS, Pereira MCA, Maciel NC, Sá RC, Araujo AD, Kruel G, Andrade LR, Pereira OJ (2019) Restinga net. Available at: http://www.restinga.net. Accessed 8 Apr 2019
Archak S, Gaikwad AB, Gautam D, Rao EVVB, Swamy KRM, Karihaloo JL (2003a) Comparative assessment of DNA fingerprinting techniques (RAPD, ISSR and AFLP) for genetic analysis of cashew (Anacardium occidentale L.) accessions of India. Genome 46:362–369
Archak S, Gaikwad AB, Gautam D, Rao EVVB, Swamy KRM, Karihaloo JL (2003b) DNA fingerprinting of Indian cashew (Anacardium occidentale L.) varieties using RAPD and ISSR techniques. Euphytica 230:397–404
Archak S, Gaikwad AB, Swamy KRM, Karihaloo JL (2009) Genetic analysis and historical perspective of cashew (Anacardium occidentale L.) introduction into India. Genome 52:222–230
Barros LM (1991) Caracterização morfológica e isoenzimática do cajueiro (Anacardium occidentale L.) tipos comum e anão-precoce, por meio de técnicas multivariadas. PhD Thesis, Universidade de São Paulo, Piracicaba
Bhattacharya A (2004) Flower visitors and fruit set of Anacardium occidentale. Ann Bot Fenn 41:385–392
Bionoset (2019) BIONOSET: Biodiversity of Piauí, Ceará and Maranhão. Available at: http://bionoset.myspecies.info/node/64. Accessed 13 Jul 2019
Borges ANC (2015) Caracterização genética em germoplasmo de cajuí (Anacardium spp.) por meio de marcadores morfoagronômicos e moleculares ISSR. MSc Thesis, Universidade Federal do Piauí, Teresina
Borges ANC, Lopes ACA, Britto FB, Vasconcelos LFL, Lima PSC (2018) Genetic diversity in a cajuí (Anacardium spp.) germplasm bank as determined by ISSR markers. Genet Molec Res 17:1–14. https://doi.org/10.4238/gmr18212
Carmo TVB, Martins LSS, Musser RS, Silva MM, Santos JPO (2017) Genetic diversity in accessions of Passiflora cincinnata Mast. based on morphoagronomic descriptors and molecular markers. Revista Caatinga 30:68–77. https://doi.org/10.1590/1983-21252017v30n108rc
Cavalcanti JJV, Wilkinson MJ (2007) The first genetic maps of cashew (Anacardium occidentale L.). Euphytica 157:131–143
Cota LG, Moreira PA, Brandão MM, Royo VA, Melo Junior AF, Menezes EV, Oliveira DA (2017) Structure and genetic diversity of Anacardium humile (Anacardiaceae): a tropical shrub. Genet Molec Res 16:1–13. https://doi.org/10.4238/gmr16039778
Crawford LA, Koscinski D, Keyghobadi N (2012) A call for more transparent reporting of error rates: the quality of AFLP data in ecological and evolutionary research. Molec Ecol 21:5911–5917
Crespo MFV, Souza LI (2014) Cajuí: boas práticas e manejo sustentável. Editora Sieart, Comissão Ilha Ativa, Parnaíba
Croxford AE, Robson M, Wilkinson MJ (2006) Characterization and PCR multiplexing of polymorphic microsatellite loci in cashew (Anacardium occidentale L.) and their cross-species utilization. Molec Ecol Notes 6:249–251
Dasmohapatra R, Rath S, Pradhan B, Rout GR (2014) Molecular and agromorphological assessment of cashew (Anacardium occidentale L.) genotypes of India. J Appl Hort 16:215–221
Desai AR (2008) Molecular diversity and phenotyping of selected cashew genotypes of Goa, and physiological response of cv. GOA-1 to in situ moisture conservation. PhD Thesis, University of Agricultural Sciences, Dharwad
Dhanaraj AL, Bhaskara Rao EVV, Swamy KRM, Bhat MG, Teertha Prasad D, Sondur SN (2002) Using RAPDs to assess the diversity in Indian cashew (Anacardium occidentale L.) germplasm. J Hort Sci Biotechnol 77:41–47
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Earl DA, Vonholdt BM (2012) Structure Harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resources 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molec Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Fernandes AG, Lopes AS, Silva EV, Conceição GM, Araújo MFV (1996) IV. Componentes Biológicos: Vegetação. In: Fundação CEPRO (eds) Macrozoneamento costeiro do Estado do Piauí. Fundação Rio Parnaíba, Teresina, pp 43–72
Freitas B, Paxton R (1998) A comparison of two pollinators: the introduced honey bee Apis mellifera and an indigenous bee Centris tarsata on cashew Anacardium occidentale in its native range of NE Brasil. J Appl Ecol 35:109–121. https://doi.org/10.1046/j.1365-2664.1998.00278.x
Gomes MFC (2017) Diversidade e estrutura genética em populações de Anacardium spp. do Parque Nacional de Sete Cidades (PI) por meio de marcadores ISSR. MSc Thesis, Universidade Federal do Piauí, Teresina
Google Earth (2019) Google Earth Pro version 7.1.8.3036 (32-bit), build date 17 Jan 2017
Guedes CCF, Giannini PCF, Sawakuchi AO, Dewitt R, Aguiar VAP (2017) Weakening of the northeast trade winds during the Heinrich stadial 1 event recorded by dune field stabilization in tropical Brazil. Quatern Res 88:369–381
Hammer O, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Paleontol Electronica 4:1–9
Jena RA, Samal KS, Pal A, Das BK, Chand PK (2016) Genetic diversity among some promising Indian local selections and hybrids of cashew nut based on morphometric and molecular markers. Int J Fruit Sci 16:69–93
Johnson DV (1972) The cashew of northeast Brazil: a geographical study of a tropical tree crop. PhD Thesis, University of California, Los Angeles
Johnson DV (1973) The botany, origin and spread of cashew (Anacardium occidentale L.). J Plantation Crops 1:1–7
Kell S, Maxted N, Frese L, Iriondo JM, Ford-Lloyd BV, Kristiansen K, Katsiosis A, Teeling C, Branca F (2012) In situ conservation of crop wild relatives: a methodology for identifying priority genetic reserve sites. In: Maxted N, Dulloo ME, Ford-Lloyd BV, Frese L, Iriondo JM, Carvalho MAAP (eds) Agrobiodiversity conservation: securing the diversity of crop wild relatives and landraces. CABI Publishing, Wallingford, pp 7–19
Kumar J, Agrawal V (2017) Analysis of genetic diversity and population genetic structure in Simarouba glauca DC. (an important bio-energy crop) employing ISSR and SRAP markers. Industr Crops Prod 100:198–207
Lima VPMS (1986) Fruteiras: uma opção para o reflorestamento do Nordeste. BNB/ETENE, Fortaleza
Luz CLS, Mitchell JD, Pirani JR, Pell SK (2019) Anacardiaceae in Flora do Brasil 2020 under construction. Jardim Botânico do Rio de Janeiro. Available at: http://floradobrasil.jbrj.gov.br/reflora/floradobrasil/FB4380. Accessed 9 Mar 2019
Martins S, Simões F, Matos J, Silva AP, Carnide V (2014) Genetic relationship among wild, landraces and cultivars of hazelnut (Corylus avellana) from Portugal revealed through ISSR and AFLP markers. Pl Syst Evol 300:1035–1046
Mitchell JD, Mori SA (1987) The cashew and its relatives (Anacardium: Anacardiaceae). Mem New York Bot Gard 42:1–76
Mneney EE, Mantell SH, Bennet M (2001) Use of random amplified polymorphic DNA (RAPD) markers to reveal genetic diversity within and between populations of cashew (Anacardium occidentale L.). J Hort Sci Biotechnol 76:375–383. https://doi.org/10.1080/14620316.2001.11511380
Mohana GS, Vanitha K, Savadi S (2018) Annual report 2017–2018. ICAR-Directorate of Cashew Research, Puttur
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nunes CF, Ferreira JL, Generoso AL, Dias MSC, Pasqual M, Cançado GMA (2013) The genetic diversity of strawberry (Fragaria ananassa Duch.) hybrids based on ISSR markers. Acta Sci Agron 35:443–452. https://doi.org/10.4025/actasciagron.v35i4.16737
Oliveira NNS, Viana AP, Quintal SSR, Paiva CL, Marinho CS (2014) Análise de distância genética entre acessos do gênero Psidium via marcadores ISSR. Revista Brasil Frutic 36:917–923. https://doi.org/10.1590/0100-2945-413/13
Paulino FDG (1992). Polinizacao entomófila em cajueiro (Anacardium occidentale L.) no litoral de Pacajús - CE. MSc Thesis, Universidade de São Paulo (ESALQ), Piracicaba
Peakall R, Smouse PE (2012a) GenAlEx 6.5: Genetic analysis in Excel: population genetic software for teaching and research – an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Peakall R, Smouse PE (2012b) GenAlEx Tutorial 2: Genetic distance and analysis of molecular variance (AMOVA). Available at: https://biology-assets.anu.edu.au/GenAlEx/Tutorials.html. Accessed 14 Jul 2019
Peakall R, Smouse PE (2015) GenAlEx 6.502 Download and Documentation [Sep 10, 2015]; Appendix 1 – Methods and Statistics in GenAlEx 6.5. Available at: https://biology-assets.anu.edu.au/GenAlEx/Download.html. Accessed 5 Mar 2019
Pessoni L (2007) Estratégias de análise da diversidade em germoplasma de cajueiro (Anacardium spp. L.). PhD Thesis, Universidade Federal de Viçosa, Viçosa
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Wen X, Falush D (2010) Documentation for structure software: Version 2.3, 2 February 2010. Available at: https://web.stanford.edu/group/pritchardlab/structure_software/release_versions/v2.3.4/html/structure.html. Accessed 19 Feb 2019
Ribeiro EKMD, Rêgo MMC, Machado ICS (2008) Cargas polínicas de abelhas polinizadores de Byrsonima chrysophylla Kunth (Malpighiaceae): fidelidade e fontes alternativas de recursos florais. Acta Bot Brasil 22:165–171. https://doi.org/10.1590/S0102-33062008000100017
Rodrigues JF, Van Den Berg C, Abreu AG, Novello M, Veasey EA, Oliveira CX, Koehler S (2015) Species delimitation of Cattleya coccinea and C. mantiqueirae (Orchidaceae): insights from phylogenetic and population genetics analyses. Pl Syst Evol 301:1345–1359. https://doi.org/10.1007/s00606-014-1156-z
Rout GR, Samal S, Nayak S, Nanda RM, Lenka PC, Das P (2002) An alternative method of plant DNA extraction of cashew (Anacardium occidentale L.) for randomly amplified polymorphic DNA (RAPD) analysis. Gartenbauwissenschaft 67:114–118
Rufino MSM (2004) Qualidade e potencial de utilização de cajuís (Anacardium spp.) oriundos da vegetação litorânea do Piauí. MSc Thesis, Universidade Federal do Piauí, Teresina
Rufino MSM, Corrêa MPF, Alves RE, Barros LM, Leite LAS (2007) Suporte tecnológico para a exploração racional do cajuizeiro. Embrapa Agroindústria Tropical, Fortaleza
Rufino MSM, Corrêa MPF, Alves RE, Leite LAS, Santos FJS (2008) Utilização atual do cajuí nativo da vegetação litorânea do Piauí, Brasil. Proc Interamer Soc Trop Hort 52:147–149
Samal S, Lenka PC, Nanda RM, Nayak S, Rout GR, Das P (2002) Genetic relatedness in cashew (Anacardium occidentale L) germplasm collections as determined by randomly amplified polymorphic DNA. Genet Resources Crop Evol 51:161–166
Samal S, Rout GR, Lenka PC (2003) Analysis of genetic relationships between populations of cashew (Anacardium occidentale L.) by using morphological characterisation and RAPD markers. Pl Soil Environm 49:176–182
Santos-Filho FS, Almeida EB Jr, Soares CJRS, Zickel CS (2010) Fisionomias das restingas do Delta do Parnaíba, Nordeste, Brasil. Revista Brasil Geogr Física 3:218–227
Sethi K (2015) Studies on morphological and molecular diversity in cashew (Anacardium occidentale L.) hybrids. PhD Thesis, Orissa University of Agriculture and Technology, Bhubaneswar
Silva AVC, Freire KCS, Lédo AS, Rabbani ARC (2014) Diversity and genetic structure of Brazilian accesses of genipapo (Genipa americana L.). Sci Agric (Piracicaba) 71:387–393. https://doi.org/10.1590/0103-9016-2014-0038
Thimmappaiah, Santhosh WG, Shobha GS, Melwyn GS (2009) Assessment of genetic diversity in cashew germplasm using RAPD and ISSR markers. Sci Hort 120:411–417
USAID-BRASIL (2006) Análise da indústria de castanha de caju: inserção de micro e pequenas empresas no mercado internacional, vol. 1. USAID, São Paulo
Vašek J, Čepková PH, Viehmannová I, Ocelák M, Huansi DC, Vejl P (2017). Dealing with AFLP genotyping errors to reveal genetic structure in Plukenetia volubilis (Euphorbiaceae) in the Peruvian Amazon. PLoS One 12:1–24. e0184259 https://doi.org/10.1371/journal.pone.0184259
Vieira M, Mayo SJ, Andrade IM (2014) Geometric morphometrics of leaves of Anacardium microcarpum Ducke and A. occidentale L. (Anacardiaceae) from the coastal region of Piauí, Brazil. Braz J Bot 37:315–327. https://doi.org/10.1007/s40415-014-0072-3
Whitlock R, Hipperson H, Thompson DBA, Butlin RK, Burke T (2016) Consequences of in situ strategies for the conservation of plant genetic diversity. Biol Conservation 203:134–142. https://doi.org/10.1016/j.biocon.2016.08.006
Wu W, Chen F, Yeh K, Chen J (2019) ISSR analysis of genetic diversity and structure of plum varieties cultivated in southern China. Biology 8:1–13. https://doi.org/10.3390/biology8010002
Acknowledgements
Thanks are due to the Parnaíba Municipal Prefecture for financial support via Mac-Doubles Fernandes do Nascimento de Apoio a Ciência, Tecnologia e Inovação programme, and to the System for Authorization and Information on Biodiversity (Instituto Chico Mendes de Conservação da Biodiversidade). The final author is grateful to the Federal University of Piauí for a research productivity grant (UFPI/PROPESQ - PRPG – 01/2018) from the Programa de Bolsa de Produtividade em Pesquisa. S.J. Mayo thanks the Royal Botanic Gardens Kew for infrastructural support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Christian Parisod.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Information on Electronic Supplementary Material
Information on Electronic Supplementary Material
Online Resource 1. Oligonucleotides of the ISSR molecular markers tested.
Online Resource 2. Images of three selected gels showing electrophoresis runs.
Online Resource 3. Binary data matrix of ISSR markers configured for GenAlEx 6.502.
Online Resource 4. UPGMA dendrogram of populations using Nei’s genetic distance.
Online Resource 5. Multiple comparisons of ΦPT fixation index and P values between populations.
Online Resource 6. Multiple comparisons of Nei’s genetic distance and identity between populations.
Online Resource 7. Bivariate plot showing lack of correlation between inter-population molecular dissimilarity (ΦPT) and geographical distance.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
dos Santos, J.O., Mayo, S.J., Bittencourt, C.B. et al. Genetic diversity in wild populations of the restinga ecotype of the cashew (Anacardium occidentale) in coastal Piauí, Brazil. Plant Syst Evol 305, 913–924 (2019). https://doi.org/10.1007/s00606-019-01611-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-019-01611-4