
Abstract
The modification of graphite screen-printed electrodes (SPEs) is reported using an eco-friendly and extremely fast method based on the direct cobalt pin electrode-to-SPE spark discharge at ambient conditions. This approach does not utilize any liquids or chemical templates, does not produce any waste, and allows the in-situ generation of CoxOy nanoparticles onto the electrode surface and the development of efficient electrocatalytic sensing surfaces for the determination of H2O2. Co-spark SPEs were characterized using scanning electron microscopy, energy-dispersive X-ray spectroscopy and x-ray photoelectron spectroscopy (XPS), revealing the formation of surface confined CoxOy nanoparticles and the diverse oxidation states of cobalt species. Co-spark SPEs were also characterized with cyclic voltammetry and electrochemical impedance spectroscopy. Redox transitions of the surface confined electrocatalysts are demonstrated by electrochemical polarization studies, showing the formation of different oxides (CoxOy), varying the XPS results. Amperometric measurements at 0.3 V vs. Ag/AgCl revealed a linear relationship between the current response and the concentration of H2O2 over the range 1 − 102 μM, achieving a limit of detection (3σ/m) of 0.6 μM. The interference effect of various electroactive species was effectively addressed by employing dual measurements in the absence and presence of the enzyme catalase. The analytical utility of the method was evaluated in antioxidant rich real-world samples, such as energy drinks, demonstrating sufficient recovery.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Energy drinks is a group of carbonated beverages that has grown significantly popular over the past two decades. Their basic ingredients are caffeine (up to 0.04% w/v), carbon dioxide and other components such as sugars, salts, taurine, amino acids and B-complex water-soluble vitamins [1]. They also contain various reducing species to suppress the oxidation process caused by oxygen, oxygen derived species (ROS) and free radicals [2]. While the adverse effects of ROS and free radicals are usually regulated by multiple protective responses in vivo, the chemistry of mixtures of redox-active ingredients and oxygen is relatively unregulated in formulated food products, particularly aqueous beverages, such as energy drinks [2, 3]. In the presence of oxygen and any oxidisable compounds acting as substrates, hydrogen peroxide (H2O2) can be generated by progressive reduction of molecular oxygen, and consequently, there is the possibility of its ingestion, in the case of beverages and foods. Over the past years there have been reports of the production of H2O2 in various foods and drinks. The production of H2O2 has been verified in beer brewing, originating from L-cysteine and thiol-rich proteins respectively [4]. Another instance is the production of H2O2 in polyphenolic beverages like cocoa [5], green [6,7,8,9] and black tea [7, 9], red wine [8], and similar phenolic-rich drinks under physiological conditions.
The non-enzymatic electrochemical determination of H2O2 can be achieved through its oxidation at (noble) metal electrodes at high overpotentials (> + 0.65 V versus common reference electrodes) [10]. However, this method may lead to a loss of selectivity in the presence of other reducing species and fouling phenomena, gradually reducing the response of the electrodes [10]. Alternatively, H2O2 can be reduced under sufficiently cathodic potentials, typically in deoxygenated solutions to mitigate interference from the oxygen reduction reaction [9].
To address these limitations, a significant amount of research has been dedicated to the modification of electrode surfaces with various charge-transfer mediators [11,12,13], electrocatalysts [14,15,16], conducting polymers [17, 18], biomolecules [19,20,21], noble metal (such as platinum, and gold) nanoparticles (NPs) [9, 22,23,24] etc. The current emphasis is on the advancement of chemical sensors employing non-noble (like copper, nickel, iron, and cobalt) metal NPs, particularly through eco-friendly methods that eliminate the need for organic solvents and costly reagents. This approach aligns with the principles of green chemistry and is geared towards reducing sensor costs [25,26,27,28,29,30].
Among the non-precious metal based electrocatalysts for H2O2, spinel type (A2+B23+X42−, where A2+and B23+ are metal cations and X42− are usually chalcogens such as oxygen [31,32,33,34] or sulfur [35, 36]), nano structures of ferromagnetic elements (Fe, Co and Ni), which present redox pairs that can effectively mediate the electro oxidation or reduction of H2O2, have been also proposed [31, 33,34,35,36].
Our study details the in-situ modification of graphite screen-printed electrodes (SPEs) using cobalt oxide nanoparticles (CoxOy NPs) through an eco-friendly spark-discharge process. This process occurs between a cobalt pin electrode and the graphite SPE, leading to instant surface modification and advanced electrocatalytic properties towards H2O2. The direct modification of the electrode surface with spark-generated nanoparticles represents a robust approach in line with environmental sustainability considerations and the principles of green chemistry in modern electroanalysis [30]. This method has been acknowledged as highly effective for developing various sensing surfaces (such as Mo NPs [37], Au NPs [38, 39], Ag NPs [40], carbon NPs and nanosheets [41,42,43]) with a simple, liquid-free, and cost-effective procedure. The effective modification of graphite SPEs with CoxOy NPs is substantiated through scanning electron microscopy (SEM), x-ray photoelectron spectroscopy (XPS), cyclic voltammetry, and electrochemical impedance spectroscopy (EIS). Furthermore, the analytical efficacy of Co-spark SPEs for the determination of H2O2 in energy drinks is demonstrated.
Experimental
Materials
A cobalt piece (Sigma-Aldrich, 99.5% trace metal basis) was fine cut with a metal saw to narrow strips to be used as cathode material (electrode pin) in the sparking process. Before use, the strips were thoroughly rinsed and sonicated with acetone. Sodium hydroxide, sodium dihydrogen phosphate, potassium chloride, ascorbic acid, caffeine and D-glucose were purchased from Merck. Hexaammineruthenium(III) chloride (RuHex) was purchased from Aldrich. Catalase from bovine liver (EC 1.11.1.6, ≥ 200 KU mL−1) was a Fluka product. A stock solution of ca. 0.1 M H2O2 was prepared by appropriate dilution of the stock product (30% H2O2, Supelco) in double distilled water (DDW), stored at 4 °C, and was weekly standardized with the permanganate method. Working solutions were daily prepared by appropriate dilutions of the stock solution in DDW.
Apparatus
Electrochemical measurements were conducted with an Autolab PGSTAT12/FRAII electrochemical analyser (Metrohm Autolab) in a conventional 3 − electrode cell. Plain or cobalt sparked SPEs (Co-spark SPEs) were used as the working electrode, while a Ag/AgCl 3 M KCl electrode (IJ Cambria) and a platinum wire served as the reference and the counter electrode, respectively. All the potential values quoted are referred to the potential of the reference electrode.
Cyclic voltammograms (CVs) were recorded in 0.1, 0.5 or 1 M NaOH at a scan rate of 0.05 V s−1 (unless stated otherwise). EIS spectra were recorded in 0.5 M NaOH over the frequency range from 100 kHz to 0.1 Hz using a sinusoidal excitation signal of 10 mV (rms) amplitude superimposed on a DC potential of 0.120 V or 0.500 V. Amperometry measurements were conducted in stirred (300 r.p.m) solutions of 0.5 M NaOH at 0.3 V. XPS measurements were conducted under ultrahigh vacuum with a base pressure of 2 × 10−9 mbar using a SPECS GmbH instrument equipped with a monochromatic MgKa source (hv = 1253.6 eV) and a Phoibos-100 hemispherical analyzer. The energy resolution was set to 1.18 eV and the photoelectron take-off angle was 45° with respect to the surface normal. Recorded spectra were set with energy step set of 0.05 eV and dwell time of 1 s. All binding energies were referenced with regard to the C1s core level centered at 284.6 eV. Spectral analysis included a Shirley background subtraction and peak deconvolution involved mixed Gaussian–Lorentzian functions was conducted with a least squares curve-fitting program (WinSpec, University of Namur, Belgium). Field-emission scanning electron microscopy (FE-SEM) images were taken with a Phenom Pharos G2 desktop FEG-SEM (ThermoFisher Scientific) at 11 kV on chromium coated samples (Quorum Q150T ES plus, sputter coater).
Fabrication and modification of electrode surface
The in-situ modification of the graphite SPE surface with spark-generated CoxOy NPs was implemented using a 16 × "linear" sparking mode. This involved connecting the cobalt electrode pin as the cathode ( −) and the graphite SPE as the anode ( +) to a high-voltage power supply. The two electrodes were brought into proximity (approximately 1 mm) through a G-code-controlled 2D positioning device until spark discharge occurred at 1.2 kV DC under ambient conditions. An external capacitor (2.8 nF) was connected in parallel to the power supply output terminals. Details on the experimental setup for electrode modification with electrical discharge and the fabrication of the graphite SPE can be found in Refs. [38, 41, 42] and Ref. [40], respectively. The electroactive area (A) of the plain and Co-spark SPE was calculated using double potential step chronocoulometry in 1 mM RuHex in 0.1 M KCl according to the procedure given in Ref. [44].
Analytical procedure
Energy drink samples were purchased at the local market. The samples were degassed in an ultrasonication bath for 10 min, and then were used to prepare the following solutions: (A) 1.0 mL sample, 0.5 mL 1 M phosphate buffered saline (PBS) pH 6, and 0.5 mL 2 M NaOH; (B) 1.0 mL spiked sample (950 μL sample plus 50 μL 50 mM H2O2), 0.5 mL 1 M PBS pH 6, and 0.5 mL 2 M NaOH; (C) 1.0 mL sample, 0.5 mL enzyme solution (480 μL 1 M PBS pH 6 plus 20 μL catalase), and, after the hand mixing of the solution for 10 min, 0.5 mL 2 M NaOH. In sample (C), PBS pH 6, is used to maximize the enzymatic activity of catalase [45], while in samples (A) and (B), which do not contain catalase, PBS was added to ensure that the same assay protocol was applied for all the measurements. Catalase was employed to eliminate H2O2, and as a result, the signal of solution (C) is ascribed to the electroactive species coexisting in the sample. Consequently, it was subtracted from the signals of solutions (A) and (B).
Amperometric measurements were conducted under stirring in an electrochemical cell containing a 2.0 mL aliquot as described for solutions A, B, or C, and 8.0 mL 0.5 M NaOH. The concentration of H2O2 in both the plain and spiked samples was determined by applying the standard addition method.
Results and discussion
Morphological studies
SEM images of plain and Co-spark SPE are shown in Fig. 1A and B, respectively. It is apparent that while the plain SPE has a compact layered structure of graphite, the Co-spark SPE shows exfoliated, micrometre-sized graphite sheets enriched with spherical cobalt nanoparticles. This double impact of the spark process benefits the sparked electrode in terms of sensitivity due to the electrocatalytic effect steaming from the cobalt-based NPs and the augmentation of the electroactive surface area of the SPE due to exfoliated nanosheets produced. Based on chronocoulometric measurements in 1 mM RuHex in 0.1 M KCl, the electroactive area of plain and Co-sparked SPEs [44], was found to be 0.1126 cm2 and 0.1883 cm2, respectively. From the EDS mapping shown in Figs. 1C-E, the spark generated nanoparticles exhibit both cobalt (depicted with red colour in Fig. 1D) and oxygen (depicted with green colour in Fig. 1E) sites which demonstrate the formation of CoxOy NPs on the electrode surface. The average particle diameter of CoxOy NPs was found to be 163 ± 73 nm.

SEM images of (A) plain and (B) Co-spark SPE. EDS mapping of (C) carbon, (D) cobalt and (E) oxygen atoms on the surface of Co-spark SPE
XPS studies
XPS studies were conducted in the surface of a Co-spark SPE (Figs. 2A, C) and a Co-spark SPE after its treatment with five cyclic voltammetry scans between 0.0 and 0.7 V in 0.5 M NaOH (termed Co-spark SPE-NaOH) (Figs. 2B, D). The Co \(2p\) spectrum involves two main peaks corresponding to \(2{p}_{1/2}\) and \(2{p}_{3/2}\) spin orbitals. The binding energies of \(2{p}_{1/2}\) and \(2{p}_{3/2}\) are separated by 15.3 eV at Co-spark SPE and 15.8 eV at Co-spark SPE-NaOH, indicating a difference of 0.5 eV for the two electrodes. At the Co-spark SPE, the \(2{p}_{3/2}\) orbit is deconvoluted into two peaks at 781.2 eV and 782.9 eV corresponding to the presence of Co(III) and Co(II) species, respectively. The existence of the intense shake-up satellite located at 787.0 eV is attributed to the high-spin nature of Co(II) species [46]. In accordance with previous works [46,47,48], the observed peaks and positions indicate the formation of Co3O4 spinel structure.

The Co \(2p\) spectrum of (A) Co-spark SPE and (B) Co-spark SPE after five cyclic voltammetry scans from 0 to 0.7 V in 0.5 M NaOH. The C \(1s\) spectrum of (C) Co-spark SPE and (D) Co-spark SPE after five cyclic voltammetry scans from 0 to 0.7 V in 0.5 M NaOH
In the case of Co-spark SPE-NaOH, shown in Fig. 2B, the two fitted peaks of the \(2{p}_{3/2}\) orbit are shifted to higher binding energies (781.6 and 783.1 eV). The shift of the binding energies for the Co-spark SPE-NaOH in combination with the difference of the energy separation of about 0.5 eV between the \(2{p}_{1/2}\) and \(2{p}_{3/2}\) orbitals at the two sparked electrodes as well as the lack of the shake-up features in the spectrum of Co-spark SPE-NaOH lead to the conclusion that the high-spin phase of Co(II) is present only at the Co-spark SPE. Conversely, we can deduce that both the peaks at 781.6 and 783.1 eV are attributed to low-spin Co(III) species [47]. However, it is evident that the Co(III) peak at 781.6 eV in \(2{p}_{3/2}\) orbit at Co-spark SPE-NaOH can be attributed to the same Co(III) species existing also in Co-spark SPE, while the peak at 783.1 eV can be attributed to CoOOH, that is, the product of the OH− adsorption and simultaneous oxidation of CoO (782.9 eV in \(2{p}_{3/2}\) orbit at Co-spark SPE) according to the chemical equation, \(CoO+{OH}^{-}\rightleftharpoons CoOOH+{e}^{-}\).
The C1s photoelectron peak is deconvoluted into four peaks at Co-spark SPE and three peaks at Co-spark SPE-NaOH, respectively as shown in Fig. 2C, D. The basic carbon frame consists of C − C/C = C bonds, while at lower binding energies, at both samples, a small peak which is attributed to the C − Co bond can also be seen. The formation of the C − Co bond can be explained considering the extremely high temperatures, up to 20000 K [49], grown locally due to the sparking process (XPS spots have been selected on the sparked areas).
Electrochemical characterization
Figure 3A shows the cyclic voltammetric behavior of Co-spark SPE within the potential window from 0 to 0.7 V in 0.5 M NaOH. The recorded CV exhibits two pairs of peaks, which both correspond to quasi-reversible redox transitions. The first redox transition is manifested by a pair of well-defined peaks with a formal potential of ca. 0.12 V, which can be attributed to the following equation:
while the second redox transition is manifested by a pair of broad peaks centered at ca. 0.5 V. According to previous works, this pair of peaks can either be attributed to the complete oxidation of Co(III) to Co(IV) according to Eq. 2 [50, 51]:
or to the adsorption of hydroxyl species and the anodic dissolution of the anodic layer [50, 51]. In our case, the fact that no Co(IV)O2 species were identified in the XPS characterization of the Co-spark electrode after the CV treatment in NaOH, urges us to lean towards the hydroxyl species adsorption explanation.

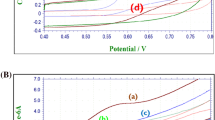
A Cyclic voltammogram of Co-spark SPE in 0.5 M NaOH. Scan rate, 50 mV s−1. B Nyquist plots of Co-spark SPE in 0.5 M NaOH at 0.12 V and C 0.5 V. Inset graphs illustrate the respective equivalent electrical circuits depicted at the same coloration
The effect of scan rate on the cyclic voltammetric behavior of Co-spark SPE was examined with CV measurements at different scan rates from 10 to 500 mV s−1 (Fig. S1A). As can be seen in Fig. S1B the peak current values for the first pair of peaks (Ipa1, Ipc1) have a linear relationship to the square root of the scan rate, indicating a diffusion-limited electrochemical process [52]. Considering that Co3(II,III)O4 molecules are confined onto the electrode, the formation of three molecules Co(III)OOH from one Co3(II,III)O4 molecule through a diffusion-limited electrochemical process (Eq. 1) can be explained as follows: Co3(II,III)O4, is a mixed oxide incorporating the Co(II)O and Co2(III)O3 forms. Co(II)O is oxidized to Co(III)OOH through an 1e−/OH− mechanism, while at the same time, through the transfer of a H2O molecule, one molecule of Co2(III)O3 forms two Co(III)OOH molecules [53]. Thus, the CoOOH formation is dependent on the mass transfer (diffusion) of hydroxyl ions and water molecules from the solution to the electrode. Acknowledging the challenge of accurately measuring the current at the second pair of (broad) peaks (Fig. S1A), linear plots between Ipa2 and Ipc2 with the square root of the scan rate were also received (Fig. S1C), suggesting a diffusion-limited electrochemical process [52]. However, based on the XPS data indicating that Co(IV)O2 is not formed, the mechanism of this redox transition may be more complex than described by Eq. 2.
In response to the cyclic voltammetric behavior of the Co-spark SPE in alkaline conditions, EIS studies were also conducted by applying either a DC potential of 0.12 V (the formal potential of the Co3O4/CoOOH redox couple) or 0.5 V (the formal potential of the second redox transition). When the impedance measurements were conducted at 0.12 V and under alkaline conditions, the impedance spectrum (Fig. 3B) exhibited a distorted semicircle over the high frequency range followed by a straight line over the low frequency range. Based on previous studies by Lyons and Brandon [54] regarding the impedimetric behavior of oxide-covered Ni, Co, and Fe electrodes, the obtained impedimetric data were effectively modeled using the equivalent electrical circuit shown as an inset graph in Fig. 3B. The circuit is represented as R1(QfRf) (Qdl[Rct(CaW)]), where R1 represents the electrolyte resistance, (QfRf) represent the dielectric properties of the Co3O4 film [50], Qdl represents the capacitance of the double-layer, Rct represents the charge transfer resistance of the redox transition (Eq. 1), and (CaZW) [55] represents the coupled hydroxyl ions diffusion and adsorption, modeling the relaxation of the charge associated with the adsorbed intermediate of the CoOOH phase. As evident from the slope of the linear part of the spectrum over the low-frequency range (slope ≠ 1), the acquired impedance cannot be solely attributed to the semi-infinite diffusion of hydroxyl anions modeled by the Warburg impedance (ZW). Instead, it is indicative of a coupled diffusion/adsorption process modeled by ZW and Ca components connected in parallel [55].
On the other hand, when the impedance measurements were conducted at 0.5 V and under alkaline conditions, the impedance spectrum illustrated in Fig. 3C can be sufficiently modeled with a quite similar equivalent electrical circuit (Fig. 3C, inset graph). In this case, the (QfRf) time constant is not included, which can be interpreted as the complete transition of Co3O4 to other CoxOy species at this potential. Consequently, the final equivalent circuit is R1(Qdl[Rct(CaZW)]), where all the symbols have their aforementioned meaning.
Optimization studies
The electrocatalytic activity of the “linear’’ mode Co-spark SPEs towards the electro oxidation of H2O2 was studied by comparing the cyclic voltammetric responses of SPEs modified with a different number of sparking lines (12, 14, 16, 18, and 20) in the absence and the presence of 5 mM H2O2. The mean electrocatalytic response and the standard deviation of the measurements with three different electrodes in each case is illustrated in Fig. 4. The highest electrocatalytic responses were observed for a modification of 16 lines, and therefore, Co-spark SPEs modified with 16 sparking lines were selected for subsequent work.

Electrocatalytic currents of Co-spark SPEs modified by a different number of sparking lines (12 − 20) in 0.5 M NaOH, containing 5 mM H2O2. Errors bars represent the standard deviation of the measurements at three different electrodes
The effect of the electrolyte on the electrocatalytic activity of Co-spark SPE was also investigated. As shown in Fig. 5, CVs of Co-spark SPE were recorded in 0.1 M phosphate buffered saline (PBS) at pH 7 and 0.1 M NaOH in the absence (dashed line) and presence (solid line) of 5 mM H2O2. The data revealed a poor electrocatalytic response in neutral pH, while the remarkable electrocatalytic behaviour in alkaline pH seems to be related to the first redox transition Co3O4/CoOOH and the chemical reduction of the electrochemically generated CoOOH by H2O2 to Co3O4, according to Eq. 3. This is then re-oxidized to CoOOH during the sweep (Eq. 1), giving rise to a several-fold increase in the anodic current, while the cathodic current decreases accordingly.

Cyclic voltammograms of Co-spark SPEs in (black line) PBS pH 7 and (red line) 0.1 M NaOH in the (dashed line) absence and (solid line) presence of 5 mM H2O2. Scan rate 50 mV s−1
Consequently, it stands to reason that since the electrocatalytic reaction is mediated by CoOOH species, the concentration of NaOH plays a large role to the electrocatalytic process. The optimum concentration of NaOH was determined by examining the cyclic voltammetry response at 0.1, 0.5, and 1 M NaOH, as illustrated in Fig. S2. In both cases of 0.5 and 1 M NaOH there is a shift of the first redox transition Co3O4/CoOOH to lower potentials (ca. 0.2 to 0.12 V), compared with that in 0.1 M NaOH, since in those cases there is an abundance of hydroxyl ions facilitating the formation of CoOOH at lower overpotentials. This shift is also prevalent in the presence of 5 mM H2O2, which favours its electrocatalysis at lower overpotentials as well. However, while both 0.5 and 1 M NaOH enhance the electrocatalytic activity of the modified electrode, the faradaic current produced in the case of 0.5 M NaOH is higher. Therefore, the concentration of 0.5 M NaOH was chosen as the optimum.
Calibration features
The amperometric response of Co-spark SPEs at various concentrations of H2O2 over the range 1 − 102 μM at three different polarization voltages was investigated. The amperograms at 0.1, 0.2, and 0.3 V and the respective calibration plots are shown in Fig. 6, while the major electroanalytical performance parameters are listed in Table 1. Based on these data and judging by the sensitivity and the limit of detection (LOD), calculated as 3Sa/slope, at each case, the polarization voltage of 0.3 V was selected as optimum for subsequent work on the determination of H2O2 in real-world samples.

(Left panel) Amperometric plots of Co-spark SPEs over the concentration range 1 – 102 μM Η2Ο2 at (black line) 0.1, (blue line) 0.2, and (red line) 0.3 V in 0.5 M NaOH. (Right panel) The corresponding calibration plots
The repeatability of the measurements for three successive additions of 10 μM H2O2 was found to be 2.9%, while the inter-electrode reproducibility among four different Co-spark SPEs at the concentration level 10 μM H2O2 was found to be 5.6%. The storage stability of the sensor was also evaluated on a weekly basis by measuring a standard of 10 μM H2O2, and it was found that Co-spark SPEs maintained more than 85% of their original signal over the course of one month.
Compared with other works on the electrochemical determination of H2O2 listed in Table 2, Co-spark SPEs exhibit favourable [31, 56,57,58,59, 61,62,63] or comparable [60, 65] analytical features. Considering their low cost, ease of fabrication, and eco-friendliness of modification, it is believed that Co-spark SPEs are highly promising electroanalytical platforms for the determination of H2O2.
Application in real energy drink samples
The response of Co-spark SPEs to three common compounds present in energy drink like ascorbic acid, glucose, and caffeine, at a concentration of 5 μM, was investigated with amperometric measurements at 0.3 V in 0.5 M NaOH. Even though the response of Co-spark SPEs to glucose and caffeine was nil, ascorbic acid gave a significant amperometric response which hampered the determination of H2O2. The interference effect of ascorbic acid and the potential interference of other reducing compounds that might exist in real-world samples were effectively addressed by employing dual measurements in the absence and presence of the enzyme catalase. Following the assay protocol described above, the method was applied to the determination of H2O2 in two commercial energy drinks. The accuracy of the method was evaluated by recovery studies at both samples fortified with 25 μM H2O2. The concentration of H2O2 in both the unspiked and spiked samples was determined using the standard addition method (Fig. 7). The responses of Co-spark SPEs before the three additions, were corrected to that obtained in the corresponding unspiked sample in the presence of catalase. Results are shown in Table 3.

A Representative amperometric plots of Co-spark SPEs showing the response in the unspiked sample #1 containing catalase, the unspiked sample #1 plus three additions of 25 μΜ Η2Ο2, and the sample #1 spiked with 25 μΜ Η2Ο2 plus three additions of 25 μΜ Η2Ο2. Standard addition plots for the B unspiked and C spiked sample #1
Conclusions
This work employs low-cost and eco-friendly semi-disposable graphite screen-printed electrodes modified with an ease to perform, extremely fast (9 s), liquid-free method based on direct cobalt pin-to-electrode electrical discharge under ambient conditions.
SEM inspection showed that the direct sparking process has a dual effect on the electrode surface generating both low-dimensional micrometre-sized graphite sheets and spherical cobalt-based nanoparticles with an average diameter of 163 ± 73 nm. Interestingly, after the sparking process, the electroactive area of the electrodes increased by 167%, from 0.1126 to 0.1883 cm2. Based on the EDS data, the spark-generated nanoparticles represent different oxide cobalt-based species (CoxOy), which according with the XPS data can be attributed to Co3O4 spinel type nanostructures. Furthermore, XPS data also indicated the formation of C − Co bonds that probably occurred due to the extremely high temperatures grown locally due to the sparking process.
Cyclic voltammetric studies demonstrated advanced electrocatalytic properties towards the electro oxidation of H2O2 at alkaline conditions, enabling the amperometric determination of the target over the concentration range 1 − 102 μM (LOD 0.6 μM). Due to the high electrocatalytic properties of spark generated CoxOy NPs toward other reducing compounds, potential interferences in real-world samples were mitigated by subtracting the signal obtained from the sample containing catalase. The analytical data obtained from antioxidant-rich real-world samples, such as energy drinks, suggest that the method holds promise for the routine analysis of H2O2 in various food and drink products with minimal sample preparation.
Data availability
All data generated or analysed during this study are included in this published article and the supplementary information file.
References
Katsa M, Proestos C (2019) 5 - Vitamin Analysis in Juices and Nonalcoholic Beverages, in: A.M. Grumezescu, A.M.B.T.-E.T. in the B.I. Holban (Eds.), Woodhead Publishing, pp. 137–173. https://doi.org/10.1016/B978-0-12-815258-4.00005-6.
Bopitiya D, Christensen D, Martin M, Zhang J, Bennett LE (2021) Production of hydrogen peroxide in formulated beverages is associated with the presence of ascorbic acid combined with selected redox-active functional ingredients. Food Chem 338:127947. https://doi.org/10.1016/j.foodchem.2020.127947
Bopitiya D, Guo S, Hearn MTW, Zhang J, Bennett LE (2022) Formulations of selected Energy beverages promote pro-oxidant effects of ascorbic acid and long-term stability of hydrogen peroxide. Food Chem 388:133037. https://doi.org/10.1016/j.foodchem.2022.133037
Muller R (1997) The formation of hydrogen peroxide during oxidation of thiol-containing proteins. J Inst Brew 103:307–310. https://doi.org/10.1002/j.2050-0416.1997.tb00961.x
Long LH, Lan ANB, Hsuan FTY, Halliwell B (1999) Generation of hydrogen peroxide by “Antioxidant” beverages and the effect of milk addition. Is cocoa the best beverage? Free Radic Res 31:67–71. https://doi.org/10.1080/10715769900300611
Ayabe S, Aoshima H (2007) Aqueous extract of citrus peel reduces production of hydrogen peroxide in catechin-enriched green tea. Food Chem 104:1594–1598. https://doi.org/10.1016/j.foodchem.2007.03.009
Akagawa M, Shigemitsu T, Suyama K (2003) Production of hydrogen peroxide by polyphenols and polyphenol-rich beverages under quasi-physiological conditions. Biosci Biotechnol Biochem 67:2632–2640. https://doi.org/10.1271/bbb.67.2632
Chai PC, Long LH, Halliwell B (2003) Contribution of hydrogen peroxide to the cytotoxicity of green tea and red wines. Biochem Biophys Res Commun 304:650–654. https://doi.org/10.1016/S0006-291X(03)00655-7
Gatselou VA, Giokas DL, Vlessidis AG, Prodromidis MI (2015) Rhodium nanoparticle-modified screen-printed graphite electrodes for the determination of hydrogen peroxide in tea extracts in the presence of oxygen. Talanta 134:482–487. https://doi.org/10.1016/j.talanta.2014.11.033
Gorton L (1995) Carbon paste electrodes modified with enzymes, tissues, and cells. Electroanalysis 7:23–45. https://doi.org/10.1002/elan.1140070104
Ricci F, Palleschi G (2005) Sensor and biosensor preparation, optimisation and applications of Prussian Blue modified electrodes. Biosens Bioelectron 21:389–407. https://doi.org/10.1016/j.bios.2004.12.001
Karyakin AA (2001) Prussian blue and its analogues: electrochemistry and analytical applications. Electroanalysis 13:813–819. https://doi.org/10.1002/1521-4109(200106)13:10%3c813::AID-ELAN813%3e3.0.CO;2-Z
Tsiafoulis CG, Trikalitis PN, Prodromidis MI (2005) Synthesis, characterization and performance of vanadium hexacyanoferrate as electrocatalyst of H2O2. Electrochem Commun 7:1398–1404. https://doi.org/10.1016/j.elecom.2005.10.001
Prieto-Simón B, Armatas GS, Pomonis PJ, Nanos CG, Prodromidis MI (2004) Metal-dispersed xerogel-based composite films for the development of interference free oxidase-based biosensors. Chem Mater 16:1026–1034. https://doi.org/10.1021/cm035110u
Sun N, Guan L, Shi Z, Li N, Gu Z, Zhu Z, Li M, Shao Y (2006) Ferrocene peapod modified electrodes: preparation, characterization, and mediation of H2O2. Anal Chem 78:6050–6057. https://doi.org/10.1021/ac060396i
Wang T, Zhu H, Zhuo J, Zhu Z, Papakonstantinou P, Lubarsky G, Lin J, Li M (2013) Biosensor based on ultrasmall MoS2 nanoparticles for electrochemical detection of H2O2 Released by cells at the nanomolar level. Anal Chem 85:10289–10295. https://doi.org/10.1021/ac402114c
Raffa D, Leung KT, Battaglini F (2003) A Microelectrochemical enzyme transistor based on an N-Alkylated Poly(Aniline) and its application to determine hydrogen peroxide at neutral pH. Anal Chem 75:4983–4987. https://doi.org/10.1021/ac0341620
Li G, Wang Y, Xu H (2007) A hydrogen peroxide sensor prepared by electropolymerization of pyrrole based on screen-printed carbon paste electrodes. Sensors 7:239–250. https://doi.org/10.3390/s7030239
Song H, Ni Y, Kokot S (2013) A novel electrochemical biosensor based on the hemin-graphene nano-sheets and gold nano-particles hybrid film for the analysis of hydrogen peroxide. Anal Chim Acta 788:24–31. https://doi.org/10.1016/j.aca.2013.06.016
Bensana A, Achi F, Bouguettoucha A, Chebli D (2019) Amperometric determination of hydrogen peroxide and its mathematical simulation for horseradish peroxidase immobilized on a sonogel carbon electrode. Anal Lett 52:1215–1235. https://doi.org/10.1080/00032719.2018.1528614
Chen W, Cai S, Ren Q-Q, Wen W, Zhao Y-D (2012) Recent advances in electrochemical sensing for hydrogen peroxide: a review. Analyst 137:49–58. https://doi.org/10.1039/C1AN15738H
Pingarrón JM, Yáñez-Sedeño P, González-Cortés A (2008) Gold nanoparticle-based electrochemical biosensors. Electrochim Acta 53:5848–5866. https://doi.org/10.1016/j.electacta.2008.03.005
Siangproh W, Dungchai W, Rattanarat P, Chailapakul O (2011) Nanoparticle-based electrochemical detection in conventional and miniaturized systems and their bioanalytical applications: a review. Anal Chim Acta 690:10–25. https://doi.org/10.1016/j.aca.2011.01.054
Chen S, Yuan R, Chai Y, Hu F (2013) Electrochemical sensing of hydrogen peroxide using metal nanoparticles: a review. Microchim Acta 180:15–32. https://doi.org/10.1007/s00604-012-0904-4
Daemi S, Ghasemi S, Akbar Ashkarran A (2019) Electrospun CuO-ZnO nanohybrid: tuning the nanostructure for improved amperometric detection of hydrogen peroxide as a non-enzymatic sensor. J. Colloid Interface Sci 550:180–189. https://doi.org/10.1016/j.jcis.2019.04.091
Cai J, Xu W, Liu Y, Zhu Z, Liu G, Ding W, Wang G, Wang H, Luo Y (2019) Robust Construction of Flexible Bacterial Cellulose@Ni(OH)2 paper: Toward High Capacitance and Sensitive H2O2 Detection. Eng Sci 5:21–29. https://doi.org/10.30919/es8d669
Li Q, Gao W, Zhang X, Liu H, Dou M, Zhang Z, Wang F (2018) Mesoporous NiO nanosphere: a sensitive strain sensor for determination of hydrogen peroxide. RSC Adv 8:13401–13407. https://doi.org/10.1039/C8RA01313F
Medhi A, Kumar Giri M, Mohanta D (2022) Non-enzymatic electrochemical detection of H2O2 using Ni (OH)2 nanoparticles. Mater Today Proc 68:262–267. https://doi.org/10.1016/j.matpr.2022.09.497
Dhara K, Mahapatra DR (2019) Recent advances in electrochemical nonenzymatic hydrogen peroxide sensors based on nanomaterials: a review. J Mater Sci 54:12319–12357. https://doi.org/10.1007/s10853-019-03750-y
Yáñez-Sedeño P, Campuzano S, Pingarrón JM (2020) (Bio)electroanalysis in the field of greener analytical chemistry. In: Garrigues S, de la Guardia M (Eds.), Challenges Green Anal Chem, The Royal Society of Chemistry, p. 0. https://doi.org/10.1039/9781788016148-00181.
Ensafi AA, Alinajafi HA, Jafari-Asl M, Rezaei B, Ghazaei F (2016) Cobalt ferrite nanoparticles decorated on exfoliated graphene oxide, application for amperometric determination of NADH and H2O2. Mater Sci Eng C 60:276–284. https://doi.org/10.1016/j.msec.2015.11.053
Chen C-H, Chen Y-C, Lin M-S (2013) Amperometric determination of NADH with Co3O4 nanosheet modifiedelectrode. Biosens Bioelectron 42:379–384. https://doi.org/10.1016/j.bios.2012.10.086
Kogularasu S, Govindasamy M, Chen S-M, Akilarasan M, Mani V (2017) 3D graphene oxide-cobalt oxide polyhedrons for highly sensitive non-enzymatic electrochemical determination of hydrogen peroxide. Sensors Actuators B Chem 253:773–783. https://doi.org/10.1016/j.snb.2017.06.172
Venosta L, Bracamonte MV, Rodríguez MC, Jacobo SE, Bercoff PG (2017) Comparative studies of hybrid functional materials based on different carbon structures decorated with nano-magnetite. Suitable application as platforms for enzyme-free electrochemical sensing of hydrogen peroxide. Sensors Actuators B Chem 248:460–469. https://doi.org/10.1016/j.snb.2017.03.159
Ramesh A, Ajith A, Gudipati NS, Vanjari SR, John SA, Biju V, Subrahmanyam C (2023) Hybridization of Co3S4 and graphitic carbon nitride nanosheets for high-performance nonenzymatic sensing of H2O2. Biosensors 13. https://doi.org/10.3390/bios13010108
Naderi L, Shahrokhian S, Amini MK, HafeziKahnamouei M (2023) Comparison of electrocatalytic performance of CuCo2O4 nanorods and nanospheres decorated with Co3S4 nanosheets for electrochemical sensing of hydrogen peroxide and glucose in human serum. ACS Appl Nano Mater 6:2755–2769. https://doi.org/10.1021/acsanm.2c05164
Kolozof P-A, Florou AB, Spyrou K, Hrbac J, Prodromidis MI (2020) In-situ tailoring of the electrocatalytic properties of screen-printed graphite electrodes with sparked generated molybdenum nanoparticles for the simultaneous voltammetric determination of sunset yellow and tartrazine. Sensors Actuators B Chem 304:127268. https://doi.org/10.1016/j.snb.2019.127268
Trachioti MG, Tzianni EI, Riman D, Jurmanova J, Prodromidis MI, Hrbac J (2019) Extended coverage of screen-printed graphite electrodes by spark discharge produced gold nanoparticles with a 3D positioning device. Assessment of sparking voltage-time characteristics to develop sensors with advanced electrocatalytic properties. Electrochim Acta 304:292–300. https://doi.org/10.1016/j.electacta.2019.03.004
Trachioti MG, Karantzalis AE, Hrbac J, Prodromidis MI (2019) Low-cost screen-printed sensors on-demand: Instantly prepared sparked gold nanoparticles from eutectic Au/Si alloy for the determination of arsenic at the sub-ppb level. Sensors Actuators B Chem 281:273–280. https://doi.org/10.1016/j.snb.2018.10.112
Papavasileiou AV, Hoder T, Medek T, Prodromidis MI, Hrbac J (2023) Sensitive riboflavin sensing using silver nanoparticles deposited onto screen-printed electrodes via controlled-energy spark discharges. Talanta 258:124409. https://doi.org/10.1016/j.talanta.2023.124409
Trachioti MG, Hemzal D, Hrbac J, Prodromidis MI (2020) Generation of graphite nanomaterials from pencil leads with the aid of a 3D positioning sparking device: application to the voltammetric determination of nitroaromatic explosives. Sensors Actuators B Chem 310:127871. https://doi.org/10.1016/j.snb.2020.127871
Trachioti MG, Hrbac J, Prodromidis MI (2021) Determination of 8−hydroxy−2ˊ−deoxyguanosine in urine with “linear” mode sparked graphite screen-printed electrodes. Electrochim Acta 399:139371. https://doi.org/10.1016/j.electacta.2021.139371
Papavasileiou AV, Trachioti MG, Hrbac J, Prodromidis MI (2022) Simultaneous determination of guanine and adenine in human saliva with graphite sparked screen-printed electrodes. Talanta 239:123119. https://doi.org/10.1016/j.talanta.2021.123119
Trachioti MG, Lazanas AC, Prodromidis MI (2023) Shedding light on the calculation of electrode electroactive area and heterogeneous electron transfer rate constants at graphite screen-printed electrodes. Microchim Acta 190:251. https://doi.org/10.1007/s00604-023-05832-w
Lück H (1965) Catalase. In: H.-U.B.T.-M. of E.A. Bergmeyer (Ed.), Academic Press, 885–894. https://doi.org/10.1016/B978-0-12-395630-9.50158-4.
Frost DC, McDowell CA, Woolsey IS (1972) Evidence for multiplet splitting of 2p photoelectron lines of transition metal complexes. Chem Phys Lett 17:320–323. https://doi.org/10.1016/0009-2614(72)87086-6
McIntyre NS, Cook MG (1975) X-ray photoelectron studies on some oxides and hydroxides of cobalt, nickel, and copper. Anal Chem 47:2208–2213. https://doi.org/10.1021/ac60363a034
Konkena B, Kalapu C, Kaur H, Holzinger A, Geaney H, Nicolosi V, Scanlon MD, Coleman JN (2023) Cobalt oxide 2D nanosheets formed at a polarized liquid|liquid interface toward high-performance Li-Ion and Na-Ion battery anodes. ACS Appl Mater Interfaces. https://doi.org/10.1021/acsami.3c11795
Reinmann R, Akram M (1997) Temporal investigation of a fast spark discharge in chemically inert gases. J Phys D Appl Phys 30:1125. https://doi.org/10.1088/0022-3727/30/7/010
Kessler T, Visintin A, de Chialvo MR, Triaca WE, Arvia AJ (1989) The development of a cobalt oxide spinel structure overlayer on cobalt electrodes: a modified electrode surface of electrocatalytic interest. J Electroanal Chem Interfacial Electrochem 261:315–329. https://doi.org/10.1016/0022-0728(89)85002-8
Boggio R, Carugati A, Trasatti S (1987) Electrochemical surface properties of Co3O4 electrodes. J Appl Electrochem 17:828–840. https://doi.org/10.1007/BF01007821
Bard AJ, Faulkner LR, White HS (2022) Electrochemical methods : Fundamentals and Applications, 3rd edn. John Wiley & Sons, Ltd., New Jersey
Longhi M, Formaro L (1999) An old workhorse of oxide investigations: new features of Co3O4. J Electroanal Chem 464:149–157. https://doi.org/10.1016/S0022-0728(99)00012-1
Lyons MEG, Brandon MP (2009) The significance of electrochemical impedance spectra recorded during active oxygen evolution for oxide covered Ni, Co and Fe electrodes in alkaline solution. J Electroanal Chem 631:62–70. https://doi.org/10.1016/j.jelechem.2009.03.019
Sluyters-Rehbach M (1994) Impedances of electrochemical systems: Terminology, nomenclature and representation - Part I: Cells with metal electrodes and liquid solutions (IUPAC Recommendations 1994). Pure Appl Chem 66:1831–1891. https://doi.org/10.1351/pac199466091831
Rui Q, Komori K, Tian Y, Liu H, Luo Y, Sakai Y (2010) Electrochemical biosensor for the detection of H2O2 from living cancer cells based on ZnO nanosheets. Anal Chim Acta 670:57–62. https://doi.org/10.1016/j.aca.2010.04.065
Kong L, Ren Z, Zheng N, Du S, Wu J, Tang J, Fu H (2015) Interconnected 1D Co3O4 nanowires on reduced graphene oxide for enzymeless H2O2 detection. Nano Res 8:469–480. https://doi.org/10.1007/s12274-014-0617-6
Lee KK, Loh PY, Sow CH, Chin WS (2013) CoOOH nanosheet electrodes: Simple fabrication for sensitive electrochemical sensing of hydrogen peroxide and hydrazine. Biosens Bioelectron 39:255–260. https://doi.org/10.1016/j.bios.2012.07.061
Ping J, Ru S, Fan K, Wu J, Ying Y (2010) Copper oxide nanoparticles and ionic liquid modified carbon electrode for the non-enzymatic electrochemical sensing of hydrogen peroxide. Microchim Acta 171:117–123. https://doi.org/10.1007/s00604-010-0420-3
Bo X, Bai J, Wang L, Guo L (2010) In situ growth of copper sulfide nanoparticles on ordered mesoporous carbon and their application as nonenzymatic amperometric sensor of hydrogen peroxide. Talanta 81:339–345. https://doi.org/10.1016/j.talanta.2009.12.007
Dong S, Xi J, Wu Y, Liu H, Fu C, Liu H, Xiao F (2015) High loading MnO2 nanowires on graphene paper: Facile electrochemical synthesis and use as flexible electrode for tracking hydrogen peroxide secretion in live cells. Anal Chim Acta 853:200–206. https://doi.org/10.1016/j.aca.2014.08.004
Wang J, Wang Z, Zhao D, Xu C (2014) Facile fabrication of nanoporous PdFe alloy for nonenzymatic electrochemical sensing of hydrogen peroxide and glucose. Anal Chim Acta 832:34–43. https://doi.org/10.1016/j.aca.2014.04.062
Dang W, Sun Y, Jiao H, Xu L, Lin M (2020) AuNPs-NH2/Cu-MOF modified glassy carbon electrode as enzyme-free electrochemical sensor detecting H2O2. J Electroanal Chem 856:113592. https://doi.org/10.1016/j.jelechem.2019.113592
Annalakshmi M, Balasubramanian P, Chen S-M, Chen T-W (2019) Enzyme-free electrocatalytic sensing of hydrogen peroxide using a glassy carbon electrode modified with cobalt nanoparticle-decorated tungsten carbide. Microchim Acta 186:265. https://doi.org/10.1007/s00604-019-3377-x
Chirizzi D, Guascito MR, Filippo E, Malitesta C, Tepore A (2016) A novel nonenzymatic amperometric hydrogen peroxide sensor based on CuO@Cu2O nanowires embedded into poly(vinyl alcohol). Talanta 147:124–131. https://doi.org/10.1016/j.talanta.2015.09.038
Funding
Open access funding provided by HEAL-Link Greece. We acknowledge support of this work by the project “Development of research infrastructures for the design, production and promotion of the quality and safety characteristics of agri-food and bio-functional products "(EV-AGRO-NUTRITION)” (MIS 5047235) which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme "Competitiveness, Entrepreneurship and Innovation" (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Author information
Authors and Affiliations
Contributions
M.S: Investigation, data collection and analysis; K.S: XPS studies; A.Ch.L: Data analysis, writing – original draft preparation; M.I.P: Conceptualization, funding acquisition, supervision, writing – review and editing. All authors, read, commented, and approved the submission of the manuscript.
Corresponding authors
Ethics declarations
Ethical Approval
Not applicable.
Conflict of interests
The authors declare no competing interests. M.I.P is an editor of this journal and recused him from all decisions about this paper. Otherwise, he declares no conflict of interest.
Additional information
Dedication
To Otto Wolfbeis in memoriam
I (Mamas Prodromidis) would like to dedicate this work to the memory of Professor Otto Wolfbeis. Otto, not only a top-ranked scientist but also a unique individual and a fantastic mentor, made a lasting impression on me. When I first met Otto in 2002, I was greatly impressed by his knowledge, his commanding personality, his kindness, and his unwavering support for all the members of his team. Now, in addition to all that, I feel extremely honoured to have had the opportunity to work with Otto in Microchimica Acta, and I would like to express my deep gratitude to this outstanding man and scientist for his tremendous support and friendship.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Siampani, M., Lazanas, A.C., Spyrou, K. et al. Eco-friendly spark-generated CoxOy nanoparticle-modified graphite screen-printed sensing surfaces for the determination of H2O2 in energy drinks. Microchim Acta 191, 150 (2024). https://doi.org/10.1007/s00604-024-06233-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-024-06233-3