
Abstract
Hepatitis B virus (HBV) and hepatitis C virus (HCV) are among the most endemic pathogens worldwide, with more than 500 million people globally currently infected with these viruses. These pathogens can cause acute and chronic hepatitis that progress to liver cirrhosis or hepatocellular carcinoma. Both viruses utilize multifaceted strategies to evade the host surveillance system and fall below the immunological radar. HBV has developed specific strategies to evade recognition by the innate immune system and is acknowledged to be a stealth virus. However, extensive research has revealed that HBV is recognized by dendritic cells (DCs) and natural killer (NK) cells. Indoleamine-2, 3-dioxygenase is an enforcer of sequential immune reactions in acute hepatitis B, and this molecule has been shown to be induced by the interaction of HBV-infected hepatocytes, DCs, and NK cells. The interleukin-28B genotype has been reported to influence HCV eradication either therapeutically or spontaneously, but the biological function of its gene product, a type-III interferon (IFN-λ3), remains to be elucidated. Human BDCA3+DCs have also been shown to be a potent producer of IFN-λ3 in HCV infection, suggesting the possibility that BDCA3+DCs could play a key role in developing therapeutic HCV vaccine. Here we review the current state of research on immune responses against HBV and HCV infection, with a specific focus on innate immunity. A comprehensive study based on clinical samples is urgently needed to improve our understanding of the immune mechanisms associated with viral control and thus to develop novel immune modulatory therapies to cure chronic HBV and HCV infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Worldwide approximately 350 million people are infected with hepatitis B virus (HBV) and 170 million with hepatitis C virus (HCV) [1, 2]. Both viruses are hepatotropic and principally non-cytopathic, but they establish persistent infection, and chronically infected patients progress seamlessly to liver cirrhosis and hepatocellular carcinoma within decades. Owing to the rapid progress in basic research on genomics and immunology, some aspects of the intercourse between the virus and host have been elucidated. However, there are still many unsolved questions regarding the key mechanisms, such as how HBV and HCV are able to evade the host’s immune surveillance system or trigger the chain of genetic derangements that ultimately lead to liver cancer.
In order to fully understand immune reactions in patients with primary HBV or HCV infection it is necessary to improve our understanding of the critical issues of virus and host interaction. Upon the host’s first encounter with the virus, the innate immune system should be triggered first, followed by the adaptive immune system consisting of multi epitope-specific CD4+ or CD8+ T cell and B cell responses. Impaired responses by either of these two immune systems or otherwise discordant transitions from innate to adaptive immunity can lead to the failure of spontaneous viral clearance of the HBV/HCV primary infection.
The first line of defense is regulated by interferon (IFN) types IFN-α/β, IFN-λ, and IFN-γ and by genes subsequently activated by IFN [IFN-stimulated genes (ISGs)], which are part of the innate immune system and induced by infected hepatocytes and by intrahepatic immune cells. There is a large difference between the liver and the blood in terms of the proportion of natural killer (NK) cells and dendritic cells (DCs) in the immune response. The percentage of NK cells in the liver also increases by more than threefold compared to the peripheral blood, accounting for one-third of the intrahepatic mononuclear cell population [3]. NK cells and DCs may be important cellular components of the intrahepatic innate immune system. NK cells can kill virus-infected hepatocytes by receptor-mediated lysis or by releasing cytotoxic granules, such as granzyme and perforin (cytopathic mechanism). In addition, they produce IFN-γ and tumor necrosis factor alpha (TNF-α), which can limit viral replication and even reduce covalently closed circular DNA (cccDNA) without liver damage (non-cytopathic mechanism) [4, 5]. DCs efficiently activate NK cells and induce adequate CD4+ and CD8+ T cell responses in HBV and HCV infection, while they also play a major role in maintaining the state of tolerance by inducing regulatory T cells (Tregs). DCs are at the center of the anti-viral immune network, and functional impairment of DCs leads to failure of viral clearance.
The focus of this review is the recent progress which has been made in the research field of innate immune responses in HBV and HCV infection.
Innate immunity in HBV infection: the role of NK cells and DCs
Following primary infection of adults with HBV, HBV-DNA remains undetectable in the serum for 4–7 weeks before an active and exponential phase of HBV replication occurs, which spreads over all hepatocytes and can lead to high viremia by the eighth to tenth week of infection. When the infection is self-limited, HBV-DNA rapidly decreases before the alanine aminotransferase (ALT) level peaks by the 12th–16th week [6]. A similar process was observed following inoculation of a chimpanzee with HBV: prior to maximum ALT level, the amount of HBV-DNA fell by more than 80 % within 2–3 weeks after the peak of viral replication [7]. This early virus control is probably sustained by non-cytopathic mechanisms [8]. As mentioned in the “Introduction”, the first line of non-cytopathic defense is considered to be regulated by IFN-α/β, IFN-λ, IFN-γ and by the ISGs. However, comprehensive gene expression analyses in a chimpanzee and woodchuck infected with HBV were unable to detect measurable IFNs or ISGs during the HBV entry and expansion phase, suggesting that HBV remains largely undetected by the innate immune system [7]. Intrahepatic HBV-specific CD8+T cells are believed to be the key players in the non-cytopathic and cytopathic clearance of HBV [6, 9].
Fisicaro et al. reported two intriguing cases of patients who successfully cleared HBV after the primary infection without any apparent elevation in ALT level [10]. Longitudinal immunological analyses revealed that NK cells are activated and produce IFN-γ prior to the increase in HBV antigen-specific CD4 or CD8 T cells [10]. IFN-γ, TNF-α, and IFN-α have been reported to be key players in the mediation of non-cytopathic HBV eradication [5, 11–13]. Of interest are the findings that IFN-γ and TNF-α were able to induce APOBEC3 deaminase, resulting in the reduction of cccDNA in hepatocytes [5, 13]. These data suggest that NK cells could play important roles in non-cytopathic HBV clearance before adaptive immune responses are fully evoked.
DCs are immune sentinels that initiate the primary immune response and harmonize the innate and adaptive immunity systems. Cytokines or IFNs produced by DCs can stimulate NK cells or activate CD4+ and CD8+ T cells. When intrahepatic mononuclear cells obtained from HBV-infected patients were cultured in vitro, blockade of plasmacytoid DC (pDC)-derived IFN-α downregulated the amounts of interleukin (IL)-12 and TNF-α released from adjacent cells [14], suggesting that liver pDCs play a pivotal role in regulating immune responses in situ. We have recently shown that pDCs are a key player in the inhibition of HBV replication by producing IFN-α upon primary HBV infection either directly or indirectly through their activation of NK cells [15]. Taken together, these studies suggest that sufficient DC activation mediates successful immune protection against HBV.
An understanding of the roles of NK cells and DCs in self-limited HBV infection through comprehensive studies utilizing clinical samples is needed to develop applicable therapeutic options in chronic HBV infection.
Indoleamine-2, 3-dioxygenase in acute HBV infection
Indoleamine-2, 3-dioxygenase (IDO), an IFN-γ or TNF-α inducible enzyme catalyzing tryptophan into kynurenine, has dual functions in infectious diseases, acting as a suppressor of intracellular pathogens and as an immune regulator [16]. We recently reported that vigorous activation of IDO in the early phase followed by sequential increase of chemokines (CXCL9, CXCL10, CXCL11) and cytokines (IFN-γ, TNF-α) is a hallmark of successful HBV clearance in patients with acute hepatitis B (Fig. 1a) [15]. In our study, following the peak level of CXCL9-11, which kept pace with the decline in ALT and HBV-DNA levels, an increase in CCL19 and CCL22 levels was observed in most of the cases of acute hepatitis B. CCL19 is chemotactic for the chemokine receptor CCR7 expressing naïve and central memory T cells, whereas CCL22 is a chemo-attractant for Th2 effector memory T cells and Tregs [17]. Such activation of CCL19 and CCL22 in the later phase of infection is involved in the replenishment of antigen-specific T cells and maintenance of immune memory. Such sequential and dynamic changes of immune parameters are rarely observed in patients with hepatic flare [15]. IDO activity is augmented by reciprocal activation of NK cells and pDCs mediated by IFN-γ and IFN-α. Enhanced levels of IDO were found to be capable of inhibiting HBV replication in hepatocytes [15]. In the same study, at the peak of ALT elevation, an inverse correlation was observed between IDO activity and ALT levels in patients with acute hepatitis B (Fig. 1b) [15]. IDO has been reported to be able to suppress the cytopathic function of cytotoxic T cells (CTLs) and NK cells or to induce Tregs [18], suggesting that it could be a counter-mechanism against severe liver damage. Therefore, IDO can be considered to be one of the key molecules activated at an early phase when it exerts dual functions, namely, suppressing HBV replication in a non-cytopathic manner and limiting excessive liver damage (Fig. 1c).

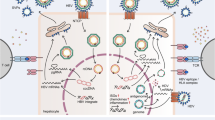
Indoleamine-2,3-dioxygenase (IDO) in hepatitis B virus (HBV) infection. a Longitudinal analyses of IDO activity and serum chemokines/cytokines in patients with acute hepatitis B (AH) and those with chronic hepatitis at hepatic flare (HF). Top panel: Left Y-axis Alanine aminotransferase (ALT) level, right Y-axis HBV-DNA. Second from top panel: Y-axis Kynurenine to tryptophan ratio (KTR) as a surrogate marker for IDO enzymatic activity. Dotted lines Average KTR level in healthy volunteers. Second from bottom panel: Left Y-axis Chemokines CXCL9 and CXCL10 level, right Y-axis chemochine CXCL11 level. Bottom panel: Left Y-axis Interferon-gamma (IFN-γ) and tumor necrosis factor (TNF) level, right Y-axis interleukin-10 (IL-10) level. HBsAg Hepatitis B surface antigen, HBcAg hepititis B core antigen, ETV entecavir. b The correlations between KTR and ALT levels in patients with AH or those at HF. Samples obtained at peak ALT level were subjected to the analyses. c Sequential activation of immune reactions enhances IDO activity at primary HBV infection (Hypothetical scheme based on the results from Yoshio et al. [15]). Firstly, dendritic cells (DC) recognize HBV infection (①). Next, CXCL9, CXCL10, and CXCL11 are induced in the liver and in turn attract DCs, natural killer (NK) cells, and T cells to the liver (②). Coexistence of NK cells and plasmacytoid DCs enhance IDO activity and suppress HBV replication in a non-cytopathic mechanism via IFN-α and IFN-γ production (③, ④). DCs produce CCL19 or CCL22 and may induce regulatory T cells (Tregs), which could alleviate liver inflammation via IL-10 production. MΦ Macrophage
Pattern recognition receptors in HBV infection
Mammalian toll-like receptors (TLRs) sense various pathogen-associated molecular patterns (PAMPs) embedded in virus components and then induce inflammatory cytokines or type-I IFN, resulting in the augmentation of anti-virus immune reactions [19]. Retinoic acid inducible gene-I (RIG-I) and melanoma differentiation antigen (MDA)-5 are cytosolic molecules that sense double-stranded (ds) RNA as a virus replicative intermediate that subsequently activates the interferon regulatory transcription factor-3 (IRF-3) and NF-kappaB (NF-κB) pathways [20]. The 5′-ε region of pregenomic HBV RNA has recently been reported to be recognizable by RIG-I in hepatocytes, which subsequently stimulates IFN-λ release [21]. RIG-I was also found to exert a direct suppressive effect on HBV replication by interfering with the binding of HBV polymerase to pregenomic RNA [21]. To date, limited information is available regarding which HBV components stimulate relevant PAMP receptors. In general, activation of TLR-dependent pathways suppresses HBV replication both in vitro and in vivo [22].
Cumulative studies have reported that the cGAS–STING (cyclic GMP-AMP synthase–Stimulator of Interferon Genes) pathway plays critical roles in the recognition of dsDNA derived from viruses, subsequently inducing IFNs and anti-viral ISGs [23]. Dansako et al. recently demonstrated that HBV-DNA is recognizable by cGAS, which stimulates STING signaling and subsequently induces ISG15 and ISG56 [24]. These ISGs are capable of suppressing the formation of HBV particles in HBV replicating hepatocytes.
Several lines of evidence have been presented that HBV or HCV impedes TLR- or RIG-I-dependent signal transduction, resulting in impaired innate immune response. In mice, hepatitis B surface antigens (HBsAg), hepatitis envelope antigen (HBeAg), and HBV virions are capable of inhibiting TLR-dependent pathways [25]. HBV-X protein directly degrades adaptor molecule MAVS, resulting in the inhibition of RIG-I-dependent signaling pathways [26]. In the shared downstream pathway of TLR and RIG-I, HBV polymerase is responsible for IRF3 inhibition at the levels of kinases TBK1/IKKε [27] and has been reported to inhibit STING-stimulated IRF3 activation and IFN-β induction [28]. HBV infection causes the inhibition of the nuclear translocation of STAT1 [29], the blockade of the TLR9–MyD88–IRAK4 axis in pDCs, or the impediment of TLR3/RIG-I signaling in hepatocytes [30]. The role of innate immunity in HBV control might be impaired for these mechanisms. The exact role of innate immunity in HBV infection needs to be investigated.
NK cells and DCs in chronic HBV infection
The NK cell response during chronic HBV infection is functionally impaired [31]. Many research groups have shown that NK cells in patients with chronic hepatitis B become defective in terms of producing IFN-γ with preserved cytotoxic activity [31–35]. Furthermore, TRAIL (TNF-related apoptosis-inducing ligand)-expressing NK cells eliminate HBV-specific T cells which highly express the TRAIL death receptor TRAIL-R2 [36]. Upon antiviral therapy, IFN-γ production by NK cells is partially restored through the downregulation of NKG2A [35].
The susceptibility of DCs to HBV is still a subject of much discussion, and the effect of HBV on DC dysfunction is known to be less than that of HCV [37, 38]. However, several reports are available on functional alterations of DC subsets in HBV infection [39], including a report of reduced pDC frequency and reduced expression of TLR9 by pDCs [40]. Liver-infiltrating pDCs are more vulnerable to HBV than their peripheral counterparts; liver pDCs have been reported to be significantly reduced and disabled in non-survivors of acute-on-chronic hepatitis B compared to those in survivors [14]. The ability of pDCs to produce IFN-α is impaired specifically in response to TLR9 but not to TLR7 agonists [41]. HBV escapes antiviral immunity by inhibiting TLR-9-mediated activation of pDCs, with consequent disruption of the interactions between pDCs and NK cells, resulting in reduced activity of NK cells in patients with chronic HBV infection [42]. Regarding the mechanisms, HBsAg is the causal factor for such disruption of the pDCs, partly by upregulating suppressor molecule SOCS-1 [41]. The dysfunction of myeloid DCs (mDCs) has also been observed, presumably due to HBV particles and HBsAg [43]. The functional impairment of NK cells and DCs in chronic HBV infection might be actively involved in viral persistency. Therefore, the role of NK cells and DCs in HBV control needs to be better elucidated.
Immunological intervention against HBV infection
Boosting virus-specific immune responses may be beneficial to HBV- or HCV-infected patients; however, critical concerns remain unsolved regarding the potential risks of evoking a severe type of hepatitis. Extensive studies using HBV transgenic mice models have been conducted. These mice are inbred to be tolerant to HBV antigens; however, in one study DCs pulsed with HBsAg induced anti-HB surface antibody (HBsAb), showing that DCs act as a tolerance breaker [44]. Active immunization by vaccine may therefore enhance innate and adaptive immunity against HBV. DCs are one of the most powerful candidates as a vehicle of viral antigens. Martinet et al. reported that peptide-loaded pDCs efficiently induced HBV-specific CD8+T cells in patients with chronic HBV infection [45]. In another study, monocyte-derived DCs retaining HBsAg recovered from chronically infected patients were able to increase the number of autologous HBV-specific T cells [46], suggesting that they are a promising adjuvant in vivo. A DNA vaccine could be another candidate: a fusion DNA encoding mouse DEC-205 linked with HBsAg successfully induced T cell and antibody responses against HBsAg in HBV transgenic mice [47]. The efficacy of a combined form of immune stimulants and viral antigens has been reported in some experimental models. Ji et al. reported that a fusion protein of T cell receptor-like antibodies (TCR-L)/IFN-α augmented an IFN-α response selectively against HBV-infected cells [48]. In mice, chimeric antigen receptors directed against HBV envelope proteins localized T cells in the liver, resulting in the reduction of HBV replication [49].
Immune stimulating reagents, such as TLR agonists, have been used to provoke generalized immune responses, with the aim to simultaneously enhance anti-HBV reactions. In one study, endogenous HBV-CpG oligodeoxynucleotides from the HBV genome stimulated IFN-α production by pDCs in a TLR-9-dependent manner, thus resulting in the clearance of HBV in HBV carrier mice [50]. The importance of TLR9 signaling has also been reported in other mice models where it induced the intrahepatic aggregates of myeloid cells and efficiently supported local proliferation of effector CD8+T cells [51]. TLR7 agonist administered orally to chimpanzees was observed to stimulate IFN-α production and the activation of ISGs and NK cells, resulting in prolonged suppression of HBV [52].
Blockade of programmed cell death (PD)-1 protein is an alternative and promising approach in chronic HBV infection. It has been reported that a combined blockade of PD-1 and CD137 in humans enhanced the responses of liver T cells against HBV [53]. In a woodchuck model with chronic hepadnaviral infection, PD-1 blockade on CD8+T cells, combined with entecavir therapy and DNA vaccination, enhanced the capacity of virus-specific T cells [54].
Innate immunity in HCV infection: HLA alleles and IL-28B/IFN-λ3
Approximately 20 % of adult patients with primary HCV infection successfully clear the HCV, while the remaining 80 % of cases fail to do so [55]. A number of host factors, such as genetic background or immune responsiveness, are now considered to be involved in dictating the distinct outcomes. Genome-wide association studies have disclosed that certain types of HLA alleles are accumulated in patients who spontaneously cleared HCV; these span from class-I to class-II haplotypes, including extended haplotypes, such as HLA-A03, -B27, -B57 and HLA-DRB1*0101, -DRB1*0401, -DRB1*1101, and -DQB1*0301 [56, 57]. It is possible that some HCV-derived peptides would be able to specifically bind to relevant HLA alleles, thus fine-tuning the direction and tone of anti-HCV immunity. Further studies aimed at exploring the functional impact of various HLA alleles or haplotypes on the pathogenesis of this disease are needed.
NK cells are one of the key players of first-line anti-viral immune responses. NK cells express various functional receptors, with one group functioning as transducing inhibitory signals [killer-cell inhibitory receptors (KIRs), CD94, NKG2A] and the other acting as activating signals (NKG2D) [12]. The function of NK cells is dynamically regulated in vivo by the balance between the expressions of counteracting receptors and their association with relevant ligands [58]. Large-scale cohort studies in HCV infection have revealed that certain combinations of HLA-C and KIR haplotypes (KIR2DL3) are closely associated with spontaneous HCV clearance [59, 60]. Such epidemiological observations raise the possibility that NK cells play an active role in HCV eradication.
Several research groups working with multiple ethnic cohorts have reported that genetic polymorphisms [single nucleotide polymorphisms (SNPs)] upstream of the promoter region of the IL-28B/IFN-λ3 gene are critically involved in the efficacy of IFN-based treatment in patients with chronic hepatitis C [61–63]. The same IL-28B SNPs have been reported to be involved in successful spontaneous HCV eradication [64]. These reports clearly indicate that IL-28B or IFN-λ3 is intricately involved in HCV eradication, but the mechanisms of their actions are still largely unknown.
Following primary exposure of the hosts to HCV, this virus maintains high replicative levels in the infected liver, resulting in the induction of IFNs and ISGs. The visualization of HCV-infected hepatocytes by in situ hybridization of the HCV genome led the authors of the study to estimate that 20–30 % of hepatocytes are infected and form clusters separated each other [65]. In the case of successful HCV eradication, it has been postulated that IFN-α/β and IFN-λ cooperatively induce anti-viral ISGs in HCV-infected hepatocytes. It is of particular interest that, in primary human hepatocytes or chimpanzee liver, IFN-λ, but not type-I IFN, is primarily induced after HCV inoculation, the degree of which is closely correlated with the levels of ISGs [66]. These results suggest that hepatic IFN-λ could be a principal driver of ISG induction in response to HCV infection.
IFN-λs, or type-III IFNs, comprise a family of highly homologous molecules consisting of IFN-λ1 (IL-29), IFN-λ2 (IL-28A), and IFN-λ3 (IL-28B). In clear contrast with type-I IFNs, they are released from relatively restricted types of cells, such as hepatocytes, intestinal epithelial cells, or DCs. Also, the cells that express hetero-dimeric IFN-λ receptors (IFN-λR1 and IL-10R2) are restricted to cells of epithelial origin, hepatocytes, or DCs [67]. Such limited profiles of cells expressing IFN-λs and their receptors define the biological uniqueness of IFN-λs. It has been shown that IFN-λs convey anti-HCV activity by inducing various ISGs [67]. Some investigators have been able to show that the expression of IL-28 in PBMCs was higher in subjects with the major IL-28B genotype than in those with the minor IL-28B genotype; however, the levels of IL-28 transcripts in liver tissue were comparable regardless of IL-28B genotype [68, 69].
DC subsets/BDCA3+DCs in acute HCV infection
Dendrite cells, as immune sentinels, sense specific genomic and/or structural components of pathogens with various pattern recognition receptors and eventually release IFNs and inflammatory cytokines [70]. In general, DCs migrate to the organ where inflammation or cellular apoptosis occurs, and they alter their function in order to alleviate or exacerbate disease conditions. Consequently, the phenotypes and/or capacity of liver DCs are affected in the inflamed liver. As mentioned in preceding sections of this review, two phenotypically and functionally distinct DC subsets have been reported in humans—myeloid DCs (mDCs) and plasmacytoid DCs (pDCs) [71]. mDCs predominantly produce IL-12 or TNF-α following exposure to pro-inflammatory stimuli, while pDCs release considerable amounts of type-I IFNs upon virus infection [71]. mDC type 2 (MDC2; BDCA3+ CD141) DCs have been drawing much attention recently since human BDCA3+DCs are reported to be a counterpart of murine CD8α+DCs [72]. Of particular interest is the report that BDCA3+DCs have a potent capacity to release IFN-λ in response to TLR3 agonist [73].
In order to explore the biological roles of BDCA3+DCs in HCV infection, our group has examined the frequency and functions of BDCA3+DCs as well as other DC subsets in patients with HCV infection. Our studies have demonstrated that human BDCA3+DCs (1) are present at an extremely low frequency in PBMCs but are accumulated in the liver (Fig. 2a, b); (2) are capable of producing IL-29/IFN-λ1, IL-28A/IFN-λ2, and IL-28B/IFN-λ3 robustly in response to HCV (Fig. 2c); (3) recognize HCV by a CD81-, endosome acidification and TRIF (TIR-domain-containing adapter-inducing interferon-β)-dependent mechanism; (4) produce larger amount of IFN-λs upon HCV stimulation in subjects with the major IL-28B genotype (rs8099917, TT) [74]. In contrast, we found that pDCs were able to produce a large amount of IFN-α/β instead of IFN-λ upon HCV infection [74, 75]. Such distinct roles of the DC subsets in response to HCV could be safeguard mechanisms of the host as protection against fatal infection. The mechanisms of increased IFN-λ3 productivity of BDCA3+DCs from subjects with the major IL-28B genotype over those with the minor genotype remain largely unknown. It has been postulated that another genetic difference might be involved in the induction of IFN-λ3, such as the length of the TATA box region. A close association between TA length and the induction of the IL-28B gene has been reported in a Japanese population [76], but not in a South East Asian population, suggesting that ethnic differences may have some effect. Other investigators have reported that the stability of IL-28B transcripts may be influenced by the specific determining region in the 3′-untranslated region of the HCV genome [77].

BDCA3+DCs in hepatitis C virus (HCV) infection. a Frequencies of BDCA3+DCs, myeloid DCs (mDCs), and plasmacytoid DCs (pDCs) in peripheral blood mononuclear cells (PBMCs) (of 21 healthy subjects) (left panel) or in the intrahepatic lymphocytes (IHLs, Liver) (of 11 HCV-negative patients who had undergone surgical resection of tumors) (right panel). Horizontal bars Mean ± standard deviation. Asterisks Significantly different at **p < 0.005 and ***p < 0.0005 by the Kruskal–Wallis test. b Paired comparisons of the frequencies of DC subsets in PBMCs and in IHLs. Results of eight patients whose PBMCs and IHLs were obtained simultaneously. Asterisks Significantly different at *p < 0.05 and ***p < 0.0005 by the paired t test. c BDCA3+DCs, pDCs, and mDC1s were cultured at 2.5 × 104 cells for 24 h with cell-cultured HCV. Results are shown as the mean + standard error of the mean from 11 experiments. Asterisks Significant at *p < 0.05, **p < 0.0005, and ***p < 0.0005 by the Kruskal–Wallis test. d Combined induction of IFN-α/β and IFN-λ from hepatocytes and DCs are crucial for the sufficient expression of liver ISGs (Scheme is from the Results section of Yoshio et al. [74]). At primary HCV infection, BDCA3+DCs and pDCs could play significant but distinct roles in the induction of IFNs and hepatic ISGs. PDCs are characterized as a potent producer of IFN-α/β. By contrast, BDCA3+DCs are characterized as a prominent producer of IFN-λs. Cooperative induction of IFN-λs and IFN-α/β from both DCs may be required for the induction of sufficient anti-HCV ISGs for HCV elimination
Only a very limited number of studies have been published on the roles of blood DCs in acute HCV infection. Ulsenheimer et al. reported that pDCs are reduced and functionally impaired in acute hepatitis C, but that this decrease is not specific to HCV infection but rather is due to liver inflammation [78]. DCs play a decisive role in shaping innate immunity by interacting with NK cells. It is tempting to speculate that the impairment of DCs in NK cell activation is partly responsible for the failure of HCV control in the early phase of primary HCV infection. Further study on the function(s) of DCs and NK cells in acute HCV infection is required for the successful design of immune therapies based on the induction of efficient antiviral responses.
IFN-λ3 in chronic HCV infection
In a study aimed at clarifying the biological significance of IFN-λ3 in patients with chronic HCV infection, we measured serum levels of IFN-λ3 in patients with chronic hepatitis C and healthy controls and found that the former had significantly higher serum IFN-λ3 levels [79]. Following HCV eradication by IFN treatment, the serum IFN-λ3 levels in the patients with chronic hepatitis C were significantly reduced, indicating that HCV infection triggers the production of IFN-λ3, a major source of which may be hepatocytes. In the same study, the serum IFN-λ3 levels were higher in patients with acute hepatitis A virus (HAV) and hepatitis E virus (HEV) infections than in healthy controls [79]. These results suggest that elevated IFN-λ levels may play protective roles in preventing the spread of hepatitis viruses. The biological functions of IFN-λ3 in patients with chronic HCV infection still need to be addressed.
DCs in chronic HCV infection
Studies on chronic HCV infection have shown that the number of mDCs and pDCs are reduced in HCV-infected patients and that both DC subtypes are impaired in their ability to promote Th1 polarization and IFN-α production [75, 80]. One of the proposed explanations for these changes effect is enhanced apoptosis in DCs, partly due to diminished NF-κB activity [81]. Dysfunctional DCs are involved in the exhaustion of CD8+ T cells, confirming that DCs play an important role in linking the innate immune system to the adaptive immune system [82] Monocyte-derived DCs generated from hepatitis C patients have been shown to have an impaired ability to stimulate allogeneic CD4 T cells [83, 84], with a decrease in the functional impairment of DCs when HCV was eradicated from patients, thereby providing evidence for HCV-induced DC disability [83]. NK cells from HCV-infected patients are able to downregulate DC functions in the presence of hepatocytes by secreting the suppressive cytokines IL-10 and transforming growth factor beta-1 [85].
Two plausible explanations for DC dysfunction in chronic HCV infection are: (1) direct infection of HCV with DCs and (2) the influence of circulating HCV proteins on the function and number of DCs. Studies involving the inoculation of cell-cultured HCV (HCVcc) into various blood cells, including DCs, have been performed in order to reveal HCV tropism [86]. However, to date no direct evidence has been obtained for the replication and reproduction of HCVcc in these cells [87–89], although HCVcc does have a negative impact on IFN-α production by pDCs, independently of direct infection of HCV [90]. Of particular interest, Dolguanic et al. proposed a novel mechanism of third-party cells involved in pDC dysfunction. These authors have shown that HCV core and NS3 trigger TNF-α production from monocytes, thereby inducing the apoptosis of pDCs and dampening their IFN-α production [91].
Further study is required to clarify whether the disability of DCs contributes to the development of HCV persistence or whether it is simply a consequence of active HCV infection.
T cell responses in chronic HCV infection
The relevance of CD4+ and CD8 T+ cells in chronic HCV infection is different from that observed with HBV. Many reports have been published on the importance of CD4+ T cell response in the clearance and control of HCV. Ulsenheimer et al. reported that in chronic hepatitis C patients, HCV-specific CD4+ T cells are functionally impaired and their activity unsustainable [92], which is in clear contrast to the situation in resolved cases. Inoculation studies of recovered chimpanzees previously infected with HCV demonstrated that CD4+ T cells were indispensable for the development of an effective CD8+ T cell response as protection from HCV persistence [93]. An increase in the number of CD161+ CCR6+ CD26+ Th17 cells capable of producing IL-21 was reported in patients who spontaneously cleared HCV [94]; the functional relevance if this result needs to be further explored.
With regard to the presence of HCV-specific CD8+ T cells during the chronic stages of disease, conflicting results have been reported regarding their roles in HCV replication and liver inflammation. HCV-specific CD8+ T cells in chronic hepatitis C patients possess a reduced capacity to proliferate and they produce less IFN-γ in response to HCV antigens [95]. Since CD8+ T cells have been reported to be involved in HCV-induced liver inflammation [96], inefficient CD8+ T cells may evoke only milder hepatocyte injury, the level of which is not sufficient for HCV eradication [97].
T cell exhaustion is a conceptual mechanism that is involved in persistency of the hepatitis virus. Under continuous exposure to a large amount of viral proteins, antigen-specific T cells become hypo-responsive to repetitive antigen stimulation in terms of proliferation and cytokine production. In the search for an inducer of exhausted T cells, extensive studies have focused on PD-1 expression on HCV-specific CD8+ T cells [98]. Urbani et al. reported that the number of CD8+ T cells expressing PD-1 increased in patients transitioning from acute hepatitis to the chronic phase [99]. However, results from other investigators observing PD-1 have been inconsistent [100]. Of particular importance, HCV-specific CD8+ T cell response is restored in vitro in the presence of masking antibodies against PD ligand 1 (PD-L1), suggesting that the PD-1/PD-L1 pathway could serve as a therapeutic target [10, 101]. In a pilot study on chimpanzees aimed at verifying the efficacy of the blockade of PD-1 using anti-PD-1 antibody against HCV infection, in one of three chimpanzees, the level of HCV-RNA was reduced during the administration of anti-PD-1 antibody, but relapsed after the cessation of treatment. A combination of PD-1 blockade with other inhibitory receptors, or the addition of direct anti-viral agents may be warranted to gain satisfactory results.
Several investigators have reported that the frequency of Tregs increase in chronic hepatitis B or C patients, either in liver or in the periphery [102, 103]. Tregs are endowed with a suppressing capacity on NK cells, DCs, and HBV- or HCV-specific T cells, thereby leading to an alleviation of collateral liver damage or impeding virus elimination. The question as to how Tregs are generated either in HBV or HCV infection is still unsolved. Extensive sequential study of intravenous drug users or healthcare workers who had higher chances of HCV infection showed that repetitive exposure to a minimal dose of HCV tended to induce Tregs, rather than raise the level of effector T cells [104]. Such regulatory mechanisms need to be avoided in order to provide sufficient immunity while awaiting a protective vaccine against HCV.
Perspectives
Synthetic compounds that specifically suppress HBV or HCV replication are now used in clinics [105]. These are quite promising therapeutic agents as an alternative approach for hard-to-treat chronic hepatitis patients, such as those infected with drug-resistant HBV strains or with compensated or decompensated liver cirrhosis. In addition to having an inhibitory effect on viral replication, such compounds are able to restore immunity either indirectly by reducing viral burden or directly by immune modulation.
In patients with chronic hepatitis B and C, a reduction of the viral load could be beneficial for the restoration of once-impaired NK cell function [106, 107]. Extensive immunological studies are needed to elucidate if the therapeutic modulation of innate immunity could shape sustained HBV- or HCV-specific adaptive immunity or exert durable maintenance of anti-tumor immune surveillance.
Most of the people living in middle or lower-income countries have poor or no access at all to costly newly developed drugs. In addition, the number of newly/repetitive HCV-infected patients is on the rise due to increasing numbers of injection drug users as a consequence of social unrest. The development of a protective/therapeutic vaccine may be one of the remedies which could help solve such socio-economic and healthcare problems. Deciphering key elements of the survival strategy of the hepatitis virus could shed light on the way to disarm such fatal and intractable pathogens.
Abbreviations
- cccDNA:
-
Covalently closed circular DNA
- CTL:
-
Cytotoxic T cell
- DC:
-
Dendritic cell
- HAV:
-
Hepatitis A virus
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HCVcc:
-
Cell-cultured HCV
- HEV:
-
Hepatitis E virus
- IDO:
-
Indoleamine-2, 3-dioxygenase
- IFN:
-
Interferon
- IRF3:
-
Interferon regulatory transcription factor-3
- ISG:
-
IFN-stimulated genes
- MDA-5:
-
Melanoma differentiation antigen-5
- mDC:
-
Myeloid dendritic cell
- NK cell:
-
Natural killer cell
- NKT:
-
Cell natural killer T cell
- PAMPs:
-
Pathogen-associated molecular patterns
- PBMCs:
-
Peripheral blood mononuclear cells
- pDC:
-
Plasmacytoid dendritic cell
- RIG-I:
-
Retinoic acid inducible gene-I
- SNPs:
-
Single nucleotide polymorphisms
- TLR:
-
Toll-like receptor
- Treg:
-
Regulatory T cell
References
Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–54.
Trepo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053–63.
Doherty DG, Norris S, Madrigal-Estebas L, et al. The human liver contains multiple populations of NK cells, T cells, and CD3+ CD56+ natural T cells with distinct cytotoxic activities and Th1, Th2, and Th0 cytokine secretion patterns. J Immunol. 1999;163:2314–21.
Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61.
Lucifora J, Xia Y, Reisinger F, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–8.
Ferrari C. HBV and the immune response. Liver Int. 2015;35[Suppl 1]:121–8.
Wieland S, Thimme R, Purcell RH, et al. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci USA. 2004;101:6669–74.
Guidotti LG, Rochford R, Chung J, et al. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–9.
Guidotti LG, Inverso D, Sironi L, et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell. 2015;161:486–500.
Fisicaro P, Valdatta C, Boni C, et al. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut. 2009;58:974–82.
Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91.
Golden-Mason L, Rosen HR. Natural killer cells: multifaceted players with key roles in hepatitis C immunity. Immunol Rev. 2013;255:68–81.
Xia Y, Stadler D, Lucifora J, et al. Interferon-gamma and tumor necrosis factor-alpha produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 2016;15:194–205.
Zhang Z, Zou ZS, Fu JL, et al. Severe dendritic cell perturbation is actively involved in the pathogenesis of acute-on-chronic hepatitis B liver failure. J Hepatol. 2008;49:396–406.
Yoshio S, Sugiyama M, Shoji H, et al. Indoleamine-2, 3-dioxygenase as an effector and an indicator of protective immune responses in patients with acute hepatitis B. Hepatology 2016;63:83–94.
Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34:137–43.
Riezu-Boj JI, Larrea E, Aldabe R, et al. Hepatitis C virus induces the expression of CCL17 and CCL22 chemokines that attract regulatory T cells to the site of infection. J Hepatol. 2011;54:422–31.
Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med. 2004;10:15–8.
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511.
Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7.
Sato S, Li K, Kameyama T, et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. 2015;42:123–32.
Guo H, Jiang D, Ma D, et al. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J Virol. 2009;83:847–58.
Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54:289–96.
Dansako H, Ueda Y, Okumura N, et al. The cyclic GMP-AMP synthetase-STING signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J 2016;283:144–56.
Wu J, Meng Z, Jiang M, et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology. 2009;49:1132–40.
Wei C, Ni C, Song T, et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol. 2010;185:1158–68.
Yu S, Chen J, Wu M, et al. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J Gen Virol. 2010;91:2080–90.
Liu Y, Li J, Chen J, et al. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J Virol. 2015;89:2287–300.
Lutgehetmann M, Bornscheuer T, Volz T, et al. Hepatitis B virus limits response of human hepatocytes to interferon-alpha in chimeric mice. Gastroenterology. 2011;140:2074–83.
Luangsay S, Gruffaz M, Isorce N, et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J Hepatol. 2015;63:1314–22.
Maini MK, Peppa D. NK cells: a double-edged sword in chronic hepatitis B virus infection. Front Immunol. 2013;4:57.
Oliviero B, Varchetta S, Paudice E, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 2009;137:1151–60.
Peppa D, Micco L, Javaid A, et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog. 2010;6:e1001227.
Schuch A, Hoh A, Thimme R. The role of natural killer cells and CD8(+) T cells in hepatitis B virus infection. Front Immunol. 2014;5:258.
Tjwa ET, van Oord GW, Hegmans JP, et al. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J Hepatol. 2011;54:209–18.
Peppa D, Gill US, Reynolds G, et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J Exp Med. 2013;210:99–114.
Lohr HF, Pingel S, Bocher WO, et al. Reduced virus specific T helper cell induction by autologous dendritic cells in patients with chronic hepatitis B—restoration by exogenous interleukin-12. Clin Exp Immunol. 2002;130:107–14.
Untergasser A, Zedler U, Langenkamp A, et al. Dendritic cells take up viral antigens but do not support the early steps of hepatitis B virus infection. Hepatology. 2006;43:539–47.
Beckebaum S, Cicinnati VR, Dworacki G, et al. Reduction in the circulating pDC1/pDC2 ratio and impaired function of ex vivo-generated DC1 in chronic hepatitis B infection. Clin Immunol. 2002;104:138–50.
Xie Q, Shen HC, Jia NN, et al. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect. 2009;11:515–23.
Xu Y, Hu Y, Shi B, et al. HBsAg inhibits TLR9-mediated activation and IFN-alpha production in plasmacytoid dendritic cells. Mol Immunol. 2009;46:2640–6.
Martinet J, Dufeu-Duchesne T, Bruder Costa J, et al. Altered functions of plasmacytoid dendritic cells and reduced cytolytic activity of natural killer cells in patients with chronic HBV infection. Gastroenterology. 2012;143(1586–1596):e8.
Op den Brouw ML, Binda RS, van Roosmalen MH, et al. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: a possible immune escape mechanism of hepatitis B virus. Immunology. 2009;126:280–9.
Akbar SM, Furukawa S, Hasebe A, et al. Production and efficacy of a dendritic cell-based therapeutic vaccine for murine chronic hepatitis B virus carrierer. Int J Mol Med. 2004;14:295–9.
Martinet J, Leroy V, Dufeu-Duchesne T, et al. Plasmacytoid dendritic cells induce efficient stimulation of antiviral immunity in the context of chronic hepatitis B virus infection. Hepatology. 2012;56:1706–18.
Gehring AJ, Haniffa M, Kennedy PT, et al. Mobilizing monocytes to cross-present circulating viral antigen in chronic infection. J Clin Invest. 2013;123:3766–76.
Yu D, Liu H, Shi S, et al. A novel dendritic-cell-targeting DNA vaccine for hepatitis B induces T cell and humoral immune responses and potentiates the antivirus activity in HBV transgenic mice. Immunol Lett. 2015;168:293–9.
Ji C, Sastry KS, Tiefenthaler G, et al. Targeted delivery of interferon-alpha to hepatitis B virus-infected cells using T-cell receptor-like antibodies. Hepatology. 2012;56:2027–38.
Krebs K, Bottinger N, Huang LR, et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145:456–65.
Lv S, Wang J, Dou S, et al. Nanoparticles encapsulating hepatitis B virus cytosine-phosphate-guanosine induce therapeutic immunity against HBV infection. Hepatology. 2014;59:385–94.
Huang LR, Wohlleber D, Reisinger F, et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol. 2013;14:574–83.
Lanford RE, Guerra B, Chavez D, et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013;144:1508–17, 1517 e1–10.
Fisicaro P, Valdatta C, Massari M, et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver T cells against HBV, but not HCV. Gastroenterology. 2012;143(1576–1585):e4.
Liu J, Zhang E, Ma Z, et al. Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog. 2014;10:e1003856.
Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36:S21–9.
Duggal P, Thio CL, Wojcik GL, et al. Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Ann Intern Med. 2013;158:235–45.
Rauch A, Gaudieri S, Thio C, et al. Host genetic determinants of spontaneous hepatitis C clearance. Pharmacogenomics. 2009;10:1819–37.
Ferlazzo G, Munz C. NK cell compartments and their activation by dendritic cells. J Immunol. 2004;172:1333–9.
Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305:872–4.
Romero V, Zuniga J, Azocar J, et al. Genetic interactions of KIR and G1 M immunoglobulin allotypes differ in obese from non-obese individuals with type 2 diabetes. Mol Immunol. 2008;45:3857–62.
Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–9.
Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401.
Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–4.
Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801.
Wieland S, Makowska Z, Campana B, et al. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology. 2014;59:2121–30.
Park HSE, Eke O, Muchmore B, Prokunina-Olsson L, Capone S, Folgori A, et al. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology 2012; 56:2060–70.
Kotenko SV. IFN-lambdas. Curr Opin Immunol. 2011;23:583–90.
Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–9.
Urban TJ, Thompson AJ, Bradrick SS, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52:1888–96.
Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–26.
Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 2001;106:259–62.
Poulin LF, Salio M, Griessinger E, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207:1261–71.
Lauterbach H, Bathke B, Gilles S, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med. 2010;207:2703–17.
Yoshio S, Kanto T, Kuroda S, et al. Human blood dendritic cell antigen 3 (BDCA3)(+) dendritic cells are a potent producer of interferon-lambda in response to hepatitis C virus. Hepatology. 2013;57:1705–15.
Takahashi K, Asabe S, Wieland S, et al. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci USA. 2010;107:7431–6.
Hiramine S, Sugiyama M, Furusyo N, et al. A thymine-adenine dinucleotide repeat polymorphism near IL28B is associated with spontaneous clearance of hepatitis C virus. J Gastroenterol. 2015;50:1069–77.
McFarland AP, Horner SM, Jarret A, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol. 2014;15:72–9.
Ulsenheimer A, Gerlach JT, Jung MC, et al. Plasmacytoid dendritic cells in acute and chronic hepatitis C virus infection. Hepatology. 2005;41:643–51.
Aoki Y, Sugiyama M, Murata K, et al. Association of serum IFN-lambda3 with inflammatory and fibrosis markers in patients with chronic hepatitis C virus infection. J Gastroenterol. 2015;50:894–902.
Kanto T, Inoue M, Miyatake H, et al. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis. 2004;190:1919–26.
Zhao L, Shields J, Tyrrell DL. Functional changes, increased apoptosis, and diminished nuclear factor-kappaB activity of myeloid dendritic cells during chronic hepatitis C infection. Hum Immunol 2010;71:751–6.
Rodrigue-Gervais IG, Rigsby H, Jouan L, et al. Dendritic cell inhibition is connected to exhaustion of CD8 + T cell polyfunctionality during chronic hepatitis C virus infection. J Immunol. 2010;184:3134–44.
Bain C, Fatmi A, Zoulim F, et al. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120:512–24.
Kanto T, Hayashi N, Takehara T, et al. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol. 1999;162:5584–91.
Jinushi M, Takehara T, Tatsumi T, et al. Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell-induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J Immunol. 2004;173:6072–81.
Liang H, Russell RS, Yonkers NL, et al. Differential effects of hepatitis C virus JFH1 on human myeloid and plasmacytoid dendritic cells. J Virol. 2009;83:5693–707.
Marukian S, Jones CT, Andrus L, et al. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 2008;48:1843–50.
Ebihara T, Shingai M, Matsumoto M, et al. Hepatitis C virus-infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology. 2008;48:48–58.
Yoon JC, Shiina M, Ahlenstiel G, et al. Natural killer cell function is intact after direct exposure to infectious hepatitis C virions. Hepatology. 2009;49:12–21.
Shiina M, Rehermann B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology. 2008;47:385–95.
Dolganiuc A, Kodys K, Kopasz A, et al. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol. 2003;170:5615–24.
Ulsenheimer A, Gerlach JT, Gruener NH, et al. Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology. 2003;37:1189–98.
Grakoui A, Shoukry NH, Woollard DJ, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302:659–62.
Kared H, Fabre T, Bedard N, et al. Galectin-9 and IL-21 mediate cross-regulation between Th17 and Treg cells during acute hepatitis C. PLoS Pathog. 2013;9:e1003422.
Wedemeyer H, He XS, Nascimbeni M, et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol. 2002;169:3447–58.
Leroy V, Vigan I, Mosnier JF, et al. Phenotypic and functional characterization of intrahepatic T lymphocytes during chronic hepatitis C. Hepatology. 2003;38:829–41.
Prezzi C, Casciaro MA, Francavilla V, et al. Virus-specific CD8(+) T cells with type 1 or type 2 cytokine profile are related to different disease activity in chronic hepatitis C virus infection. Eur J Immunol. 2001;31:894–906.
Frebel H, Richter K, Oxenius A. How chronic viral infections impact on antigen-specific T-cell responses. Eur J Immunol 2010;40:654–63.
Urbani S, Amadei B, Tola D, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–403.
Kasprowicz V, Schulze Zur Wiesch J, Kuntzen T, et al. High level of PD-1 expression on hepatitis C virus (HCV)-specific CD8+ and CD4+ T cells during acute HCV infection, irrespective of clinical outcome. J Virol. 2008;82:3154–60.
Nakamoto N, Cho H, Shaked A, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009;5:e1000313.
Stoop JN, van der Molen RG, Baan CC, et al. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41:771–8.
Sugimoto K, Ikeda F, Stadanlick J, et al. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology. 2003;38:1437–48.
Park SH, Veerapu NS, Shin EC, et al. Subinfectious hepatitis C virus exposures suppress T cell responses against subsequent acute infection. Nat Med. 2013;19:1638–42.
Forestier N, Reesink HW, Weegink CJ, et al. Antiviral activity of telaprevir (VX-950) and peginterferon alfa-2a in patients with hepatitis C. Hepatology. 2007;46:640–8.
Serti E, Chepa-Lotrea X, Kim YJ, et al. Successful interferon-free therapy of chronic hepatitis C virus infection normalizes natural killer cell function. Gastroenterology. 2015;149(190–200):e2.
Boni C, Lampertico P, Talamona L, et al. Natural killer cell phenotype modulation and natural killer/T-cell interplay in nucleos(t)ide analogue-treated hepatitis e antigen-negative patients with chronic hepatitis B. Hepatology. 2015;62:1697–709.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest to disclose.
Rights and permissions
About this article

Cite this article
Yoshio, S., Kanto, T. Host–virus interactions in hepatitis B and hepatitis C infection. J Gastroenterol 51, 409–420 (2016). https://doi.org/10.1007/s00535-016-1183-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-016-1183-3