
Abstract
Introduction
Chronic graft-versus-host disease (cGVHD) is a debilitating side effect of allogeneic hematopoietic cell transplantation (HCT), affecting the quality of life of patients. We used whole exome sequencing to identify candidate SNPs and complete a multi-marker gene-level analysis using a cohort of cGVHD( +) (N = 16) and cGVHD( −) (N = 66) HCT patients.
Methods
Saliva samples were collected from HCT patients (N = 82) pre-conditioning in a multi-center study from March 2011 to May 2018. Exome sequencing was performed and FASTQ files were processed for sequence alignments. Significant SNPs were identified by logistic regression using PLINK2v3.7 and Fisher’s exact test. One cGVHD( −) patient sample was excluded from further analysis since no SNP was present in at least 10% of the sample population. The FUMA platform’s SNP2GENE was utilized to annotate SNPs and generate a MAGMA output. Chromatin state visualization of lead SNPs was completed using Epilogos tool. FUMA’s GENE2FUNC was used to obtain gene function and tissue expression from lead genomic loci.
Results
Logistic regression classified 986 SNPs associated with cGVHD( +). SNP2GENE returned three genomic risk loci, four lead SNPs, 48 candidate SNPs, seven candidate GWAS tagged SNPs, and four mapped genes. Fisher’s exact test identified significant homozygous genotypes of four lead SNPs (p < 0.05). GENE2FUNC analysis of multi-marker SNP sets identified one positional gene set including lead SNPs for KANK1 and KDM4C and two curated gene sets including lead SNPs for PTPRD, KDM4C, and/or KANK1.
Conclusions
Our data suggest that SNPs in three genes located on chromosome 9 confer genetic susceptibility to cGVHD in HCT patients. These genes modulate STAT3 expression and phosphorylation in cancer pathogenesis. The findings may have implications in the modulation of pathways currently targeted by JAK inhibitors in cGVHD clinical trials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Each year thousands of patients suffering from blood disorders, hematological cancers, or immunodeficiency complete conditioning therapy prior to undergoing allogeneic hematopoietic stem cell transplant (HCT) in the USA [1]. In allogeneic HCT, hematopoietic cells are isolated from a related or unrelated donor and must have the same human leukocyte antigen (HLA) types as the recipient to be considered “HLA matched.” The types of HLAs typically checked are HLA-A, B, C, DRB1, and DPB1 haplotypes on chromosome 6 that are inherited [2, 3]. If no related donors with matched or partial matched HLA types are available, unrelated donors with full or partial HLA matches are preferred [3]. Those HCT patients with HLA mismatched donors (i.e., having one or more HLA type that is not identical) experience increased rates of chronic graft-versus host disease (cGVHD) and even with identical HLA matches, up to 41% of patients undergoing HCT develop cGVHD [1, 4]. When donor T cells respond to host cells, GVHD can occur [5]. cGVHD development process consists of three phases: (1) afferent phase (2) donor T cell activation, differentiation, and migration and (3) effector phase [6]. If occurring 100 days post-transplantation, GVHD is considered chronic as opposed to acute [7]. cGVHD can cause significant debilitating symptoms. Patients with cGVHD experience a lower quality of life with daily activities being impaired long after clinical symptoms of cGVHD appear. Many tissues can be affected by cGVHD including the eyes, gastrointestinal (GI) tract, liver, lungs, skin, and mouth [8].
Symptoms of cGVHD are scored on a grading scale of zero to three with zero corresponding to no organ involvement and three corresponding to the medical condition in which the function of the organ is severely compromised [9]. Furthermore, global severity of cGVHD can be described as mild, moderate, or severe. Mild cGVHD is characterized by a patient having one to two organs affected with a score of one. Moderate cGVHD is described as three or more organs affected with a score of one, at least one organ with a score of two, and a lung score of one, while severe cGVHD has at least one organ with a score of three and a lung score of two [9].
Clinical treatment of cGVHD aims to improve and to stabilize organ involvement and symptoms, but also to improve quality of life and survival [10]. Guidelines recommend no treatment or local application of steroids or other immunosuppressive drugs if mild cGVHD [11]. With moderate or severe cGVHD, the first treatment is systemic steroids. If refractory to steroids, first choice is ruxolitinib and second choice other immunosuppressive treatments such as ECP, ibrutinib, rituximab, or sirolimus [12]. The reason why some HCT patients develop more severe cGVHD is not well understood, although some studies have sought to associate genetic polymorphisms with cGVHD [13,14,15,16]. Previous studies have associated SNPs mapped to genes with cGVHD incidence including C-X-C motif chemokine receptor 3 (CXCR3), C–C motif chemokine receptor 6 (CCR6), FGFR1 oncogene partner (FGFR10P), interleukin 10 (IL10), interleukin 1 receptor type 1 (IL1R1), nuclear factor kappa B subunit 1 (NFKB1), heparanase (HSPE), and cytotoxic T-lymphocyte associated protein 4 (CTLA4) [13, 15,16,17].
Here, we carried out whole exome sequencing to (i) identify candidate single nucleotide polymorphisms (SNPs) associated with cGVHD and (ii) complete a multi-marker gene-level analysis, using a cohort of 82 allogeneic HCT patients, to better understand the underlying genetic susceptibilities distinguishing patients who developed cGVHD (N = 16) from those who did not (N = 66).
Methods
Chronic graft-versus-host disease determination
Participants were enrolled in a multicenter prospective study (OraStem) assessing oral complications related to conditioning regimens, hematopoietic cell transplantation, and immunologic reactions (mainly cGVHD) that may follow and as previously described [18]. Enrollment started at the first center in March 2011 and proceeded at intervals until May 2018. Hematological conditions treated included blood cancers or blood disorders, namely, myeloma, lymphoma, acute/chronic lymphoblastic leukemia (ALL/CLL), acute/chronic myelogenous leukemia (AML/CLL), myelodysplastic syndrome, myelofibrosis, or immune deficiency. Conditioning regimens included either non-myeloablative, myeloablative, or reduced intensity conditioning. Participants were assessed prior to the start of HCT and 100 days, 6 months, and 1 year after HCT. cGVHD was assessed prospectively on site at each follow-up visit as part of Orastem. A specific question regarding oral cGVHD was also assessed. A participant was considered positive if they were noted by the oncologist to have cGVHD at 100 days, 6 months, or 1 year after HCT. Data was not collected on grade or organ involvement of cGVHD. This study was approved by Atrium Health IRB# IRB00080071. All participants have signed an informed consent form.
Saliva sample collection, DNA isolation, library prep, and sequencing
Whole saliva was collected from adult patients from three international sites (Atrium Health Carolinas Medical Center, Charlotte, NC, USA; BC Cancer, Vancouver, BC, Canada; and two Swedish sites-Sahlgrenska University Hospital, Gothenburg and Karolinska Institute, Huddinge, Sweden) at baseline prior to conditioning therapy into Oragene collection tubes (DNA GenoTek, Ottawa, Ontario, Canada). Samples from the satellite sites were stored in the Oragene kit and were shipped at ambient temperatures to the Translational Research Laboratory at Atrium Health Carolinas Medical Center (AH-CMC), Charlotte, NC, USA. All samples were processed per manufacturer’s instructions and stored at − 80 °C until further analysis. Isolation of DNA was completed using the prepIT-L2P reagent (DNA GenoTek, Ottawa, Ontario, Canada), following manufacturer’s instructions (DNA GenoTek, Inc.).
Following DNA extraction and QC profiling, the samples were submitted for library preparation and sequencing. Libraries were generated for each sample using NEB reagents for PCR construction and PFX polymerase for the post ligation PCR. KAPA RT-PCR was completed on these libraries before hybridization. NimbleGenv3.0 hybridization reactions were completed, and resulting libraries underwent final library QC analysis which included KAPA qPCR. A Paired-end100 sequencing run was completed via the Illumina Hiseq platform.
Processing sequencing reads for SNP determination and bioinformatic analysis
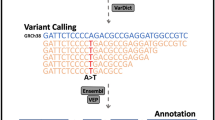
Quality control was completed using FASTQCv0.11.9 on the paired-end FASTQ files of 16 cGVHD( +) and 66 cGVHD( −) samples [19]. Adapters and low-quality reads were trimmed and paired ends were combined and aligned to the GRCh38.19 reference genome [20,21,22]. Duplicates were marked, reads were aligned, and base quality score recalibration was completed using GATKv4.2.6.1 tools [23].
Variants were called using GATKv4.2.6.1’s HaplotypeCaller [23]. Variant Call Files (VCF) were combined using BCFtoolsv1.9 and filtered using VCFtoolsv0.1.16 keeping only SNPs present in > 10% of the population with a minor allele frequency > 1% and removing multi-allelic SNPs [24, 25]. One cGVHD − patient sample was removed from downstream analysis due to not having any SNPs present in at least 10% of the sample population. Variant association analysis was then completed on filtered SNPs using PLINK2v3.7 by performing a logistic regression analysis on cGVHD + (n = 16) and cGVHD − SNPs (n = 65 of 66 samples) [26]. The genomic inflation factor of our dataset was < 1.0; therefore, covariates were not included in the logistic regression. A principal component analysis (PCA) plot of the data was created using Pythonv3.9.13 programming language packages [27,28,29].
Significant SNP positions were loaded into the Known VARiants (Kaviarv160204) online tool with GRCh38.19 used as the reference, and Reference SNP cluster IDS (RSids) were retrieved [30]. The Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA) online platform was used to annotate SNPs with the SNP2GENE tool and Multi-marker Analysis of GenoMic Annotation (MAGMA) was utilized [31]. Options included the cGVHD association logistic regression results from PLINK2v3.7 as the input, significant SNPs with RSids as the “lead snp” list (n = 909 unique out of 986 total SNPs), sample size as “N = 81,” a window of 2.5kbp upstream and 0 kbp downstream and a maximum adjusted p-value of 1 × 10−5. Manhattan plots showing the GWAS summary statistics (p < 1 × 10−5) and the gene-based test (p < 6.219 × 10−5) were created. A contingency table of observed heterozygous vs. homozygous minor alleles was then produced for cGVHD − and cGVHD + groups, and a post hoc Fisher’s exact test was used for the lead SNPs using Rv4.2.1. Chromatin state genomic visualization maps of the lead candidate SNPs were created using the Epilogos online tool with “hg38” as the human reference genome and “All 127 Roadmap Epigenomes” in the 18-state model [32].
The FUMA GENE2FUNC online tool was used to obtain gene function and tissue expression (GTExv8 54 and 30 tissue types) from SNP2GENE identified lead genomic loci (i.e., lead SNPs). Additionally, GENE2FUNC was run on all significant SNPs annotated to genes (n = 804) with recognized Ensembl IDs (n = 679; 678 unique) and all known background genes within the online tool (n = 57,241; 35,142 unique), using hypergeometric tests to determine overrepresented genes of interest (padj < 0.05).
Results
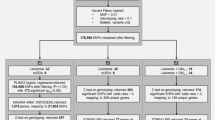
Patient demographics are presented in Table 1. Overall, analytical strategy with associated summary results is presented in Fig. 1. Filtering with VCFtoolsv0.1.16, resulted in 133,052 candidate SNPs being retained out of 1,141,608 total. One sample had to be removed from further analysis by not having any SNPs present in at least 10% of the sample population from the cGVHD( −) group resulting in 81 total patient samples used for analysis. Using PLINK2v3.7, 986 candidate SNPs from the 81 patient samples (cGVHD + , n = 16; cGVHD − , n = 65) were found significant as determined by logistic regression (Online Resource 1). A PCA plot showing the first four principal components of the cGVHD + and cGVHD − groups based on SNP data is presented in Fig. S1 (Online Resource 2). Limited clustering was observed for cGVHD − patients, while cGVHD + patients were scattered, likely due to differences in sample sizes. There were, however, no significant differences in the variation of SNPs in each group, indicating this result being independent from demographics.

Analytical design for determination of SNPs associated with cGVHD in a cohort of HCT patients by exome sequencing. Legend: Samples were collected from 82 hematopoietic cell transplant (HCT) recipients either with chronic graft-versus-host disease (cGVHD + ; N = 16) or without (cGVHD − ; N = 66). FASTQ processing was completed using Trim Galorev0.6.7, Bowtie2v2.4.1, Genome Analysis Toolkit (GATKv4.2.6.1), and BCFtoolsv1.9. VCFtoolsv0.1.16 was used to filter 1,141,582 identified single nucleotide polymorphisms (SNPs) keeping only SNPs present in > 10% of the population, a minor allele frequency of > 1%, and removing multi-allelic SNPs. Filtering resulting in 133,052 candidate SNPs for logistic regression analysis in PLINK2v3.7. PLINK2v3.7 classified 986 SNPs to be significantly associated with cGVHD + . Using the 133,052 SNPs with the 986 significant SNPs as SNP2GENE and MAGMA input, significant returned results included three genomic risk loci, four lead SNPs, 48 candidate SNPs, seven candidate GWAS tagged SNPs, and four mapped genes. Using these outcomes as input for GENE2FUNC resulted in three significant functional gene sets including one positional and two curated ones. *One cGVHD − sample was removed by not having any SNPs present in at least 10% of the sample population
Using MAGMA output in the SNP2GENE analysis, we identified three significant genomic risk loci, four lead candidate SNPs, and 48 candidate SNPs to be significantly correlated with the development of cGVHD from a possible 909 annotated SNPs among the 986 identified by logistic regression. Manhattan plots at the SNP level and gene-level analyses are presented in Fig. S2 (Online Resource 3). The four lead candidate SNPs rs7031287, rs2296055, rs7030531, and rs10405821 mapped to the genes protein tyrosine phosphatase receptor type D (PTPRD), KN motif and ankyrin repeat domains 1 (KANK1), lysine demethylase 4C (KDM4C), and paralemmin (PALM), respectively. The four lead SNPs and 48 candidate SNPs are presented in Table 2. No lead SNPs had combined annotation dependent depletion (CADD) scores greater than suggested threshold of 12.37 [33]. However, CADD scores of two candidate SNPs (rs10975820 [KANK1:R130C19.3] and rs4465021 [KDM4C]) not identified as lead SNPs, exceeded this threshold (Table 2).
The Fisher’s exact tests of the lead SNPs comparing the proportions of heterozygous and homozygous alternate genotypes resulted in three of the four lead SNPs being significant (p < 0.05) and one having a marginal significance (p = 0.0525) (Table 3 and Online Resource 4). We identified a total of 17 patients among 81, i.e., 14 in cGVHD − group and 3 in cGVHD + group with mutations in an intronic region of PTPRD (rs7031287; chr9:8,454,566: T > C). However, in the cGVHD( +) group, we identified three patient samples (18.75%) with homozygous mutations (C/C), while in the cGVHD( −) group, we observed 12 samples (18.18%) to be heterozygous and two samples (3.03%) to have a homozygous mutation (p < 0.05) (Table 3 and Online Resource 4). Genotyping of KANK1 revealed heterozygous mutations in one sample (6.25%) of the cGVHD( +) group and in 11 samples (16.66%) of the cGVHD( −) group. Furthermore, we were able to identify homozygous alternate mutations in three (18.75%) of the 16 cGVHD( +) patients and in two (3.03%) cGVHD( −) samples. Using the Fisher’s Exact test, these proportions were determined as marginally significant (p = 0.0525). Genotyping of the SNP within KDM4C (rs7030531; chr9:6,835,419:C > T) revealed heterozygous mutations in none of the cGVHD( +) samples and in 14 samples (21.21%) of the cGVHD( −) group. Furthermore, we were able to identify homozygous alternate mutations in three (18.75%) cGVHD( +) patients and in none of the cGVHD( −) samples (p < 0.05) (Table 3 and Online Resource 4). The Fisher’s exact test on genotyping of the SNP within PALM (rs10405821; chr19:731,055:C > G) revealed that the proportions of heterozygous genotype vs. homozygous genotype in our cGVHD( +) and cGVHD( −) groups were significantly different (p = 5.85 × 10−3) (Table 3 and Online Resource 4).
A heatmap of the four lead SNPs tissue expression is shown in Fig. 2. The four genes are functionally expressed in several tissues relevant to cGVHD pathology such as the skin, esophagus, small intestine, and colon. GTExv8 analysis across 54 tissue types showed PTPRD as expressed at low levels in 34 tissue types including 16 known to be affected by cGVHD (Fig. 2). KANK1 was overrepresented in 49 of 54 tissue types, 14 of which have cGVHD involvement (Fig. 2). We determined KDM4C was expressed at low levels in five tissue types involved in cGVHD including the pancreas, liver, skeletal muscle, and kidneys. Additionally, our data showed PALM being highly expressed in 48 of the GTExv8 54 tissue types including the lungs and 12 other cGVHD involved tissue types (Fig. 2).

Lead SNP genotype-tissue expression. Legend: Genotype-tissue expression (GTExv8) heatmap rendered using the Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA) online platform’s GENE2FUNC tool. The y-axis shows significant genes determined from “lead SNPs” associated with chronic graft-versus-host disease (cGVHD) from a cohort of 81 allogeneic hematopoietic stem cell transplant patients (cGVHD − , N = 65; cGVHD + , N = 16). The x-axis depicts the GTExv8 of 54 tissue types. The level of gene expression per tissue type is shown on a scale of under expressed (blue) to overexpressed (red). cGVHD tissue involvement is depicted with an asterisk (*)
Analysis performed with GENE2FUNC on the four lead SNPs resulted in one significant positional gene set (chr9p24 location) and two curated gene sets (“Davicioni molecular alveolar rhabdomyosarcoma (ARMS) vs. the embryonic form (ERMS) up” and “Snijders amplified in head and neck tumors”). The positional gene set (n = 104) contained 3 genes with significant SNPs identified by logistic regression. In addition, the genes KANK1 and KDM4C with the lead SNPs determined by SNP2GENE analysis (padj = 7.20 × 10−3), located on chromosome 9, were also members of this positional gene set as shown in Table S1 (Online Resource 5). The curated gene set “Davicioni molecular ARMS vs. ERMS up” (n = 20 significant genes of 339 genes) contained three of four lead SNPs representing KANK1, KDM4C, and PTPRD genes per SNP2GENE analysis (padj = 3.59 × 10−2) while the “Snijders amplified in head and neck tumors” gene set (n = 2 significant genes of 37 genes) contained KDM4C and PTPRD genes (padj = 3.59 × 10−2) (Online Resource 5).
The SNP in PTPRD rs7031387 (T > C) (Fig. 3) is an intron variant within an area of weak transcription in the epigenome and has a global ALFA (allele frequency aggregator) alternate allele frequency of 39%. The KANK1 gene SNP rs2296055 (T > C) is a noncoding transcription variant flanking upstream of an active transcription start site and transcription regions in the epigenome map. The ALFA alternate frequency among the general population is approximately 30% for this variant. The SNP rs7030531 (C > T) is an intron variant residing within a region of strong transcription with zinc finger genes and repeats. This SNP has an ALFA alternate frequency of 31% among the general population. The PALM SNP rs10405821, located on chromosome 19, is also an intronic SNP with an alternate allele frequency of 36% and falls downstream of an active enhancer region within a strong transcription portion of the epigenome map (Fig. 3). Visualization of the chromatin state of each lead SNP’s position of the four genes is presented in Fig. 3.


Epilogos of MAGMA identified lead SNPs. Legend: Chromatin state visualization of the lead candidate SNPs (rs7031287 a, rs2296055 b, rs7030531 c, and rs10405821 d) determined by Multi-marker Analysis of GenoMic Annotation (MAGMA) using the Epilogos online tool with “hg38” as the human reference genome and “All 127 Roadmap Epigenomes in the 18-state model. The colors represent the following chromatin states: gray
 : repressed PolyComb, light purple
: repressed PolyComb, light purple
 : Heterochromatin, pale yellow
: Heterochromatin, pale yellow
 : active enhancer, yellow
: active enhancer, yellow
 : weak enhancer, lime green
: weak enhancer, lime green
 : geneic enhancer, green
: geneic enhancer, green
 : strong transcription, dark green
: strong transcription, dark green
 : weak transcription, orange
: weak transcription, orange
 : flanking TSS upstream, red
: flanking TSS upstream, red
 : active TSS, blue
: active TSS, blue
 : ZNF genes and repeats
: ZNF genes and repeats
Discussion
This is the first study to complete a multi-marker gene-set analysis using whole exome sequencing in a cohort of allogeneic HCT patients with cGVHD. The first three SNPs were all located on chromosome 9, the fourth on chromosome 19, all belonging to specific gene sets with biological relevance.
We identified SNP rs7301287 (chr9:8,454,566:T > C) within the PTPRD gene that has been previously identified as a variant in a study investigating susceptibility to large-vessel ischemic stroke [34]. Notably, after stroke there is an acute inflammatory response with an influx of neutrophils, T cells, monocytes, and leukocytes within the brain [35, 36]. SNPs within PTPRD may also be associated with hematological cancers or blood disorders in general [37,38,39]. The SNP we identified within PTPRD had significantly more homozygous alternate alleles in the cGVHD( +) group than in the cGVHD( −) group, suggesting the homozygous genotype to constitute a risk for cGVHD. PTPRD has been classified as a predictive biomarker of immune checkpoint inhibitors in multiple cancer types including non-small cell lung cancer, skin cutaneous melanoma, and gastric cancer [40]. In this study, they found tumor mutational burden and microsatellite instability to be positively correlated with PTPRD mutations in multiple cancer types including non-small cell lung cancer, esophagogastric cancer, and colorectal cancer [40]. In head and neck squamous cell carcinoma, PTPRD is a known tumor suppressor and mutations lead to increased expression of signal transducer and activator of transcription 3 (STAT3) [41]. When STAT3 is overexpressed in immune cells, it leads to compromised immunity by inhibiting innate and adaptive immune responses, a characteristic of cGVHD [42]. It remains to be confirmed whether a change in PTPRD protein expression or the expression of a modified form contributes to cGVHD by upregulating STAT3.
The significant SNP identified within KANK1, rs2296055 (chr9:676,954:T > C), was previously identified in a study considering variants associated with coronary artery disease in an Indian population of 153 individuals [43]. Interestingly, atherosclerosis is considered primarily a chronic inflammatory condition that may be influenced by leukocyte count, interferon gamma (IFN- γ), and cytokines [44]. Additionally, another study showed that in certain cancers such as ALL KANK1 is completely deleted [45]. Although not significant, our results suggest the homozygous alternate allele (C/C) within rs2296055 may constitute a risk for cGVHD, while the heterozygous alternate allele (T/C) may be protective. Furthermore, in a study by Medves et al., the authors were able to identify a fusion of KANK1 and platelet derived growth factor receptor beta (PDGFRB) in a patient with a hematological malignancy [46]. The authors suggested that the KANK1-PDGFRB hybrid can transform hematopoietic cells and showed that STAT3 was phosphorylated in the presence of this hybrid. STAT3 can be phosphorylated by receptor-associated Janus kinases (JAK), the inhibitors of which are under investigation in clinical trials for cGVHD. Moreover, KANK1 has been discovered to fuse with neurotrophic receptor tyrosine kinase 3 (NTRK3) gene in patient samples with pathologically confirmed metanephric adenoma leading to a subset of B-Raf proto-oncogene, serine/threonine kinase (BRAF) mutations [47]. A study by Cui et al. demonstrated that KANK1 can inhibit cell growth by inducing apoptosis in malignant peripheral nerve sheath tumor cell lines [48]. The gene KANK1 also functions as a cytoskeleton regulator and has been suggested as having a role in tissue development [45]. Moreover, KANK1 has been shown to regulate the polymerization of actin by inhibiting RhoA activity, similar to the Rho family of GTPases [45], which include Rho GTPase activating protein 29 (ARHGAP29) involved in the maintenance of oral mucosal epithelium integrity [49, 50]. It is therefore conceivable that KANK1 homozygous mutations in its regulatory sequence could be responsible for impaired tissue reformation and fibrosis characteristic of the third phase of cGVHD. In addition, since KANK1 function seems to be linked to the JAK pathway and has been shown to inhibit IFN-γ to promote T cell apoptosis further investigation to determine its validity as a drug target candidate for the treatment of cGVHD is warranted [51].
The third lead SNP residing on chromosome 9 (rs7030531; chr9:6,835,419:C > T) corresponded to the gene KDM4C. No previous studies have associated rs7030531 with disease. This gene has been suggested as possessing oncogenic properties in solid state tumors as well as AML by functioning as a histone H3K9 demethylase [52]. Furthermore, KDM4 demethylases have been determined as integral for long term-maintenance of hematopoiesis using mouse models [53]. It has been suggested that KDM4C is essential to the survival of cells under stress and plays a role in kidney development by regulating autophagy [54]. Although it is rare, cases of cGVHD of the kidneys have been reported in approximately 1% of patients undergoing allogeneic HCT and the risk of chronic kidney disease is increased in long-term survivors [55, 56]. While KDM4C was shown to activate HIF1α/VEGFA signaling through the costimulatory factor STAT3, its role in cGVHD remains unclear [57].
Two genes (KANK1 and KDM4C) were a part of the only significant positional gene set (chr9p24) determined. The results suggest that for the positional gene set located on chromosome 9, the chromatin state of the relevant regions might be significantly impacted by the SNPs. Chromosome 9 houses the largest cluster of interferons in the genome and contains genes related to cell growth and regulation as well as tumor suppressors [58,59,60]. In a previous study, ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1) located on chromosome 9 was found to fuse with multiple genes which are associated with leukemia [61]. Additionally, this locus is related to diseases including but not limited to renal cell carcinoma, hepatocellular carcinoma, pancreatic carcinoma, ALL, breast cancer, obsessive–compulsive disorder, and monosomy 9p syndrome [45]. Considering chromosome 9 has been associated with leukemias and contains many interferons which are an integral part of the cGVHD pathogenesis, this positional gene set should be further explored for its role in cGVHD.
A study investigating genetic variations affecting gene expression using the Avon Longitudinal Study of Parents and Children cohort identified rs10405821 meaning this SNP could potentially affect trans gene expression [62]. It is worth mentioning, dysregulation of gene expression is associated with inflammatory pathways such as monocyte cytokine responses after lipopolysaccharide stimulation that activate toll-like receptors in children with autism disorders [63]. The SNP rs10405821 is within the gene PALM, encoding paralemmin-1 a membrane anchored cytoplasmic facing palmitoylated protein [64]. Paralemmin-1 is abundant in brain tissue but is also well expressed in cGVHD relevant tissues such as lung (Fig. 2) [64]. Paralemmin-1 is a dynamic regulator of intracellular vesicle trafficking, cell shape, and movement [39]. A study by Turk et al. found that PALM was upregulated in cell lines representative of estrogen-receptor positive breast cancers [65]. Another member of the PALM family of proteins, PALM3, was previously shown to be upregulated by lipopolysaccharide-stimulation in alveolar epithelial cell lines and to be involved in lipopolysaccharide-toll-like receptor-4 signaling in alveolar macrophages. Meanwhile, downregulation of PALM3 after lipopolysaccharide-stimulation can improve the severity of lung injury and can inhibit pro-inflammatory cytokines in human lung adenocarcinoma cells [65]. It has been suggested that lipopolysaccharide stimulation through bacterial metabolites may induce differentiation of toll-like receptor 4 which could lead to GVHD [66]. The lungs can be significantly affected in cGVHD, considering that cGVHD is considered “severe” if the patient is classified with a lung score of two or more. Whether dysregulation of PALM expression has an impact on cGVHD severity through lung injury remains a matter of investigation. This gene was the only gene with significant SNPs on chromosome 19. Chromosome 19 contains a gene region of immunoglobulin like domains that can recognize polymorphic epitopes of HLA class one molecules [67]. In patients with AML undergoing HCT, these domains have been reported to influence treatment outcomes and risk of GVHD [68]. Additionally, abnormalities in chromosome 19 have been associated with megakaryoblastic leukemias and AML [69, 70]. Interestingly, since SNPs were found in three genes (i.e., PTPRD, KANK1, KDM4C) involved in the STAT3 signaling pathway, one possible functional relationship with paralemmin-1 is that STAT3 may be palmitoylated and co-localize with intracellular vesicles which transit to the perinuclear membrane [71, 72]. More research is needed (i) to determine whether ineffectiveness of JAK inhibitors in treating cGVHD relates to such alternative path of translocation of the unphosphorylated form of STAT3 to the nucleus and (ii) to confirm the involvement of PALM polymorphisms in cGVHD risk and patient outcomes.
Davicioni molecular ARMS vs. molecular ERMS is a human gene set where genes are upregulated in ARMS compared to the ERMS class of tumors [73]. ARMS and ERMS tumors mainly affect children and young adults. ARMS tumors normally affect muscles in the legs, arms, or trunk, while ERMS affect muscles of the head, neck, or genitourinary tract [74]. The second curated gene set was “Snijders amplified in head and neck cancer tumors” where deregulation of genes in this gene set relate to developmental and differentiation pathways as well as cell misspecification in oral squamous cell carcinoma [75]. How similar biological disturbances related to these gene sets impact the development of hematological cancers or blood disorders as well as the incidence and severity of cGVHD remain unknown. Furthermore, all four lead SNPs may have an impact on the transcriptional regulation of their respective associated gene, most likely as a gain in function by stimulating STAT3 pathway. Finally, among genes involved in cGVHD susceptibility identified in previous SNP association studies it is notable that CCR6 can induce phosphorylation of STAT3 in asthma and that STAT3 is an important binding partner of FGDR1 in the development of non-small cell lung cancer [13, 76, 77].
Limitations
Although our results provide possible genomic risk loci and insights into the development of cGVHD, the sample size is a limiting factor of this analysis. Indeed, of the 16 cGVHD + patients, 9 had oral signs of GVHD (i.e., lichenoid lesion, erythematous lesion, ulceration, and swollen minor salivary glands) which would not achieve sufficient power to identify associated SNPs. Additionally, the absence of clinical data on the site of cGVHD development and ethnicity of patients due to being a multi-national study limits the scope of this study. Collection of site and ethnicity data for cGVHD along with a larger cohort of allogeneic HCT patients presenting with cGVHD are needed to better investigate susceptibility to cGVHD and to rule out significant findings being only due to other genetic factors or cancer type. Another limitation is the lower-than-expected rate of cGHVD occurrence in the present study, which might correspond to a selection bias. Furthermore, the validation of SNP effects at a functional level experimentally are imperative to identify drug targets to alleviate the long-term effects of cGVHD.
Conclusions
Our data suggest that there is genetic susceptibility to cGVHD in patients undergoing allogeneic HCT which strongly involves polymorphisms in genes located on chromosome 9, with the possibility of concerted mechanisms of action. Mutations in PTPRD, KANK1, KDM4C, and/or PALM may confer predispositions to cGVHD incidence or severity requiring confirmation and validation in future studies to aid in the future drug development for cGVHD.
Data availability
All data and accompanying scripts are available via the Translational Research Lab Github repository (https://github.com/mbeckm01/GHVD_Exome).
References
Khaddour K, Hana CK, Mewawalla P (2023) Hematopoietic Stem Cell Transplantation. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/books/NBK536951/
Spellman SR (2022) Hematology 2022-what is complete hla match in 2022? Hematology 2022(1):83–89. https://doi.org/10.1182/hematology.2022000326
Giralt S, Bishop MR (2009) Principles and overview of allogeneic hematopoietic stem cell transplantation. Cancer Treat Res 144:1–21. https://doi.org/10.1007/978-0-387-78580-6_1
Ritari J, Hyvärinen K, Koskela S, Niittyvuopio R, Nihtinen A, Salmenniemi U, Putkonen M, Volin L, Kwan T, Pastinen T, Itälä-Remes M, Partanen J (2019) Computational analysis of hla-presentation of non-synonymous recipient mismatches indicates effect on the risk of chronic graft-vs.-host disease after allogeneic hsct. Front Immunol 10:1625. https://doi.org/10.3389/fimmu.2019.01625
Ferrara JL, Levine JE, Reddy P, Holler E (2009) Graft-versus-host disease. Lancet (london, england) 373(9674):1550–1561. https://doi.org/10.1016/s0140-6736(09)60237-3
Sun Y, Tawara I, Toubai T, Reddy P (2007) Pathophysiology of acute graft-versus-host disease: recent advances. Transl Res 150(4):197–214. https://doi.org/10.1016/j.trsl.2007.06.003
Justiz Vaillant AA, Modi P, Mohammadi O (2022) Graft-Versus-Host Disease. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538235/. Accessed 9 Sept 2022
Flowers ME, Parker PM, Johnston LJ, Matos AV, Storer B, Bensinger WI, Storb R, Appelbaum FR, Forman SJ, Blume KG, Martin PJ (2002) Comparison of chronic graft-versus-host disease after transplantation of peripheral blood stem cells versus bone marrow in allogeneic recipients: long-term follow-up of a randomized trial. Blood J Am Soc Hematol 100:415–419. https://doi.org/10.1182/blood-2002-01-0011
Lee SJ (2017) Classification systems for chronic graft-versus-host disease. Blood J Am Soc Hematol 129:30–37. https://doi.org/10.1182/blood-2016-07-686642
Kostareva I, Kirgizov K, Machneva E, Ustyuzhanina N, Nifantiev N, Skvortsova Y, Shubina I, Reshetnikova V, Valiev T, Varfolomeeva S, Kiselevskiy M (2022) Novel and promising strategies for therapy of post-transplant chronic gvhd. Pharmaceuticals (basel) 15:1100. https://doi.org/10.3390/ph15091100
Penack O, Marchetti M, Ruutu T, Aljurf M, Bacigalupo A, Bonifazi F, Ciceri F, Cornelissen J, Malladi R, Duarte RF, Giebel S, Greinix H, Holler E, Lawitschka A, Mielke S, Mohty M, Arat M, Nagler A, Passweg J, Schoemans H, Socie G, Solano C, Vrhovac R, Zeiser R, Kroger N, Basak GW (2020) Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: updated consensus recommendations of the european society for blood and marrow transplantation. Lancet Haematol 7:e157–e167. https://doi.org/10.1016/s2352-3026(19)30256-x
Zeiser R, Polverelli N, Ram R, Hashmi SK, Chakraverty R, Middeke JM, Musso M, Giebel S, Uzay A, Langmuir P, Hollaender N, Gowda M, Stefanelli T, Lee SJ, Teshima T, Locatelli F, Investigators R (2021) Ruxolitinib for glucocorticoid-refractory chronic graft-versus-host disease. N Engl J Med 385:228–238. https://doi.org/10.1056/nejmoa2033122
Dai H, Rachakonda SP, Penack O, Blau IW, Blau O, Radujkovic A, Müller-Tidow C, Dreger P, Kumar R, Luft T (2021) Polymorphisms in cxcr3 ligands predict early cxcl9 recovery and severe chronic gvhd. Blood Cancer J 11(2):42. https://doi.org/10.1038/s41408-021-00434-2
Giaccone L, Faraci DG, Butera S, Lia G, Di Vito C, Gabrielli G, Cerrano M, Mariotti J, Dellacasa C, Felicetti F, Brignardello E (2021) Biomarkers for acute and chronic graft versus host disease: state of the art. Expert Rev Hematol 14(1):79–96. https://doi.org/10.1080/17474086.2021.1860001
Partanen J, Hyvärinen K, Bickeböller H, Bogunia-Kubik K, Crossland RE, Ivanova M, Perutelli F, Dressel R (2020) Review of genetic variation as a predictive biomarker for chronic graft-versus-host-disease after allogeneic stem cell transplantation. Front Immunol 11:575492. https://doi.org/10.3389/fimmu.2020.575492
Kuba A, Raida L, Mrazek F, Schneiderova P, Kriegova E, Langova K, Furst T, Furstova J, Faber E, Papajik T (2020) nfkb1 gene single-nucleotide polymorphisms: implications for graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Ann Hematol 99:609–618. https://doi.org/10.1007/s00277-020-03935-5
Martin PJ, Fan W, Storer BE, Levine DM, Zhao lP, Warren EH, Flowers ME, Lee SJ, Carpenter PA, Boeckh M, Hingorani S, Yan l, Hu Q, Preus l, Liu S, Spellman S, Zhu X, Pasquini M, Mccarthy P, Stram D., … Hansen JA (2016) Replication of associations between genetic polymorphisms and chronic graft-versus-host disease. Blood, 128(20):2450–2456. https://doi.org/10.1182/blood-2016-07-728063
Brennan MT, Hasseus B, Hovan AJ, Raber-Durlacher JE, Blijlevens NM, Huysmans MC, GarmingLegert K, Johansson JE, Moore CG, Von Bultzingslowen I (2018) Impact of oral side effects from conditioning therapy before hematopoietic stem cell transplantation: protocol for a multicenter study. Jmir Res Protoc 7:e103. https://doi.org/10.2196/resprot.8982
Andrews S (2010) fastqc: a quality control tool for high throughput sequence data [online] available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 19 Aug 2022
Krueger F, James F, Ewels P, Afyounian E, Schuster-Boeckler B (2021) felixkrueger/trimgalore: v0.6.7 - doi via zenodo (0.6.7). Zenodo. https://zenodo.org/record/5127899. Acccessed 19 Aug 2022
Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, Fulton RS, Kremitzki M, Magrini V, Markovic C, Mcgrath S, Steinberg KM, Auger K, Chow W, Collins J, Harden G, Hubbard T, Pelan S, Simpson JT, Threadgold G, Torrance J, Wood JM, Clarke L, Koren S, Boitano M, Peluso P, Li H, Chin CS, Phillippy AM, Durbin R, Wilson RK, Flicek P, Eichler EE, Church DM (2017) Evaluation of grch38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27:849–864. https://doi.org/10.1101/gr.213611.116
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Van der Auwera Ga, O’connor BD (2020) Genomics in the cloud: using docker, gatk, and wdl in terra. O’reilly media, sebastopol, ca. https://www.oreilly.com/library/view/genomics-in-the/9781491975183/
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, Mccarthy SA, Davies RM, Li H (2021) Twelve years of samtools and bcftools. Gigascience 10:giab008. https://doi.org/10.1093/gigascience/giab008
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, Depristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, Mcvean G, Durbin R, Genomes project analysis g (2011) The variant call format and vcftools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation plink: rising to the challenge of larger and richer datasets. Gigascience 4:7. https://doi.org/10.1186/s13742-015-0047-8
Python software foundation. Python language reference. Available at http://www.python.org. Accessed 16 Oct 2022
Plotly technologies inc. (2015) Collaborative data science. Montreal, qc. Available at https://plot.ly.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E (2011) Scikit-learn: machine learning in python. J Mach Learn Res 12:2825–2830
Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC (2011) Kaviar: an accessible system for testing snv novelty. Bioinformatics 27:3216–3217. https://doi.org/10.1093/bioinformatics/btr540
Watanabe K, Taskesen E, Van Bochoven A, Posthuma D (2017) Functional mapping and annotation of genetic associations with fuma. Nat Commun 8:1826. https://doi.org/10.1038/s41467-017-01261-5
Meuleman et al. Epilogos: information-theoretic navigation of multi-tissue functional genomic annotations. Available at https://epilogos.altius.org/. Accessed 20 Oct 2022
Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–315. https://doi.org/10.1038/ng.2892
Janicki PK, Eyileten C, Ruiz-Velasco V, Pordzik J, Czlonkowska A, Kurkowska-Jastrzebska I, Sugino S, Imamura Kawasawa Y, Mirowska-Guzel D, Postula M (2019) Increased burden of rare deleterious variants of the KCNQ1 gene in patients with large-vessel ischemic stroke. Mol Med Rep 19(4):3263–3272. https://doi.org/10.3892/mmr.2019.9987
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87(5):779–789. https://doi.org/10.1189/jlb.1109766
Tarkanyi G, Karadi ZN, Szabo Z, Szegedi I, Csiba L, Szapary L (2020) Relationship between leukocyte counts and large vessel occlusion in acute ischemic stroke. BMC Neurol 20(1):440. https://doi.org/10.1186/s12883-020-02017-3
Depreter B, De Moerloose B, Vandepoele K, Uyttebroeck A, Van Damme A, Terras E, Denys B, Dedeken L, Dresse MF, Van Der Werff Ten Bosch J, Hofmans M, Philippe J, Lammens T (2021) Deciphering molecular heterogeneity in pediatric aml using a cancer vs. Normal transcriptomic approach. Pediatr Res 89:1695–1705. https://doi.org/10.1038/s41390-020-01199-3
Klintman J, Appleby N, Stamatopoulos B, Ridout K, Eyre TA, Robbe P, Pascua LL, Knight SJL, Dreau H, Cabes M, Popitsch N, Ehinger M, Martin-subero JI, Campo E, Mansson R, Rossi D, Taylor JC, Vavoulis DV, Schuh A (2021) Genomic and transcriptomic correlates of richter transformation in chronic lymphocytic leukemia. Blood 137:2800–2816. https://doi.org/10.1182/blood.2020005650
Olson TL, Cheon H, Xing JC, Olson KC, Paila U, Hamele CE, Neelamraju Y, Shemo BC, Schmachtenberg M, Sundararaman SK, Toro MF, Keller CA, Farber EA, Onengut-Gumuscu S, Garrett-Bakelman FE, Hardison RC, Feith DJ, Ratan A, Loughran TP (2021) Frequent somatic tet2 mutations in chronic nk-lgl leukemia with distinct patterns of cytopenias. Blood 138:662–673. https://doi.org/10.1182/blood.2020005831
Shang X, Zhang W, Zhang X, Yu M, Liu J, Cheng Y, Cheng B (2022) ptprd/ptprt mutation as a predictive biomarker of immune checkpoint inhibitors across multiple cancer types. Front Immunol 13:991091. https://doi.org/10.3389/fimmu.2022.991091
Peyser ND, Du Y, Li H, Lui V, Xiao X, Chan TA, Grandis JR (2015) Loss-of-function ptprd mutations lead to increased stat3 activation and sensitivity to stat3 inhibition in head and neck cancer. Plos One 10:e0135750. https://doi.org/10.1371/journal.pone.0135750
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X (2020) Targeting stat3 in cancer immunotherapy. Mol Cancer 19:145. https://doi.org/10.1186/s12943-020-01258-7
Bhat KG, Guleria VS, Rastogi G, Sharma V, Sharma A (2022) Preliminary genome wide screening identifies new variants associated with coronary artery disease in indian population. Am J Transl Res 14(7):5124–5131
Stöllberger C, Finsterer J (2002) Role of infectious and immune factors in coronary and cerebrovascular arteriosclerosis. Clin Diagn Lab Immunol 9(2):207–215. https://doi.org/10.1128/cdli.9.2.207-215.2002
Kakinuma N, Zhu Y, Wang Y, Roy BC, Kiyama R (2009) Kank proteins: structure, functions and diseases. Cell Mol Life Sci 66:2651–2659. https://doi.org/10.1007/s00018-009-0038-y
Medves S, Duhoux FP, Ferrant A, Toffalini F, Ameye G, Libouton JM, Poirel HA, Demoulin JB (2010) Kank1, a candidate tumor suppressor gene, is fused to pdgfrb in an imatinib-responsive myeloid neoplasm with severe thrombocythemia. Leukemia 24(5):1052–1055. https://doi.org/10.1038/leu.2010.13
Catic A, Kurtovic-Kozaric A, Sophian A, Mazur L, Skenderi F, Hes O, Rohan S, Rakheja D, Kogan J, Pins MR (2020) Kank1-ntrk3 fusions define a subset of braf mutation negative renal metanephric adenomas. BMC Med Genet 21:202. https://doi.org/10.1186/s12881-020-01143-6
Cui Z, Shen Y, Chen KH, Mittal SK, Yang JY, Zhang G (2017) Kank1 inhibits cell growth by inducing apoptosis through regulating cxxc5 in human malignant peripheral nerve sheath tumors. Sci Rep 7:40325. https://doi.org/10.1038/srep40325
Zawislak A, Wozniak K, Agirre X, Gupta S, Kawala B, Znamirowska-Bajowska A, Grocholewicz K, Lubinski J, Prosper F, Jakubowska A (2021) Association of abca4 gene polymorphisms with cleft lip with or without cleft palate in the polish population. Int J Environ Res Public Health 18:11483. https://doi.org/10.3390/ijerph182111483
Mougeot J-LC, Mougeot FB (2019) Compositions and methods for genetic markers to identify risk of oral mucositis (U.S. patent no. 20190264265). U.S. patent and trademark office. https://rb.gy/ik0fb0. Accessed 26 Oct 2022
Wu KX, Wang XT, Hu XL, Jiang XY, Zhuang JC, Xu YZ, Lin LR, Tong ML, Yang TC, liu LL (2021) Lncrna-enst00000421645 upregulates kank1 to inhibit ifn-γ expression and promote t cell apoptosis in neurosyphilis. Front Microbiol 12:749171. https://doi.org/10.3389/fmicb.2021.749171
Xue L, Li C, Ren J, Wang Y (2021) kdm4c contributes to cytarabine resistance in acute myeloid leukemia via regulating the mir-328–3p/ccnd2 axis through malat1. Ther Adv Chronic Dis 12:2040622321997259. https://doi.org/10.1177/2040622321997259
Agger K, Nishimura K, Miyagi S, Messling JE, Rasmussen KD, Helin K (2019) The kdm4/jmjd2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 134(14):1154–1158. https://doi.org/10.1182/blood.2019000855
Pan HC, Chen YH, Fang WC, Wu VC, Sun CY (2022) Essential roles of the histone demethylase kdm4c in renal development and acute kidney injury. Int J Mol Sci 23(16):9318. https://doi.org/10.3390/ijms23169318
Fraile P, Vazquez L, Caballero D, Garcia-Cosmes P, López L, San Miguel J, Tabernero JM (2013) Chronic graft-versus-host disease of the kidney in patients with allogenic hematopoietic stem cell transplant. Eur J Haematol 91(2):129–134. https://doi.org/10.1111/ejh.12149
Sakellari I, Barbouti A, Bamichas G et al (2013) Gvhd-associated chronic kidney disease after allogeneic haematopoietic cell transplantation. Bone Marrow Transplant 48:1329–1334. https://doi.org/10.1038/bmt.2013.55
Wu X, Deng Y, Zu Y, Yin J (2022) Erratum: histone demethylase kdm4c activates hif1α/vegfa signaling through the costimulatory factor stat3 in nsclc. Am J Cancer Res 12(12):5692–5693
Humphray SJ, Oliver K, Hunt AR, Plumb RW, Loveland JE, Howe KL, Andrews TD, Searle S, Hunt SE, Scott CE, Jones MC, Ainscough R, Almeida JP, Ambrose KD, Ashwell RI, Babbage AK, Babbage S, Bagguley CL, Bailey J, Banerjee R, ... Dunham I (2004) DNA sequence and analysis of human chromosome 9. Nature 429(6990):369–374. https://doi.org/10.1038/nature02465
Dessy E, Rossi E, Berenzi A, Tironi A, Benetti A, Grigolato P (2008) Chromosome 9 instability and alterations of p16 gene in squamous cell carcinoma of the lung and in adjacent normal bronchi: fish and immunohistochemical study. Histopathology 52(4):475–482. https://doi.org/10.1111/j.1365-2559.2008.02969.x
Bohlander, s. K., dreyling, m. H., hagos, f., sveen, l., olopade, o. I., & díaz, m. O. (1994) Mapping a putative tumor suppressor gene on chromosome 9 bands p21–p22 with microdissection probes. Genomics 24(2):211–217. https://doi.org/10.1006/geno.1994.1608
de Braekeleer E, Douet-Guilbert N, Rowe D, Bown N, Morel F, Berthou C, Férec C, de Braekeleer M (2011) Abl1 fusion genes in hematological malignancies: a review. Eur J Haematol 86(5):361–371. https://doi.org/10.1111/j.1600-0609.2011.01586.x
Bryois J, Buil A, Evans DM, Kemp JP, Montgomery SB, Conrad DF, Ho KM, Ring S, Hurles M, Deloukas P, Davey Smith G, Dermitzakis ET (2014) Cis and trans effects of human genomic variants on gene expression. Plos Genet 10(7):e1004461. https://doi.org/10.1371/journal.pgen.1004461
Hughes HK, Rowland ME, Onore CE et al (2022) Dysregulated gene expression associated with inflammatory and translation pathways in activated monocytes from children with autism spectrum disorder. Transl Psychiatry 12:39. https://doi.org/10.1038/s41398-021-01766-0
Kutzleb C, Sanders G, Yamamoto R, Wang X, Lichte B, Petrasch-Parwez E, Kilimann MW (1998) Paralemmin, a prenyl-palmitoyl-anchored phosphoprotein abundant in neurons and implicated in plasma membrane dynamics and cell process formation. J Cell Biol 143(3):795–813. https://doi.org/10.1083/jcb.143.3.795
Turk CM, Fagan-Solis KD, Williams KE, Gozgit JM, Smith-Schneider S, Marconi SA, Otis CN, Crisi GM, Anderton DL, Kilimann MW, Arcaro KF (2012) Paralemmin-1 is over-expressed in estrogen-receptor positive breast cancers. Cancer Cell Int 12:17. https://doi.org/10.1186/1475-2867-12-17
Chen X, Wu X, Zhao Y, Wang G, Feng J, Li Q, Qian G (2011) A novel binding protein of single immunoglobulin il-1 receptor-related molecule: paralemmin-3. Biochem Biophys Res Commun 404:1029–1033. https://doi.org/10.1016/j.bbrc.2010.12.104
Beckman MF, Morton DS, BahraniMougeot F, Mougeot JC (2021) Allogenic stem cell transplant-associated acute graft versus host disease: a computational drug discovery text mining approach using oral and gut microbiome signatures. Support Care Cancer 29(4):1765–1779. https://doi.org/10.1007/s00520-020-05821-2
Partanen J, Hyvärinen K, Bickeböller H, Bogunia-Kubik K, Crossland RE, Ivanova M, Perutelli F, Dressel R (2020) Review of genetic variation as a predictive biomarker for chronic graft-versus-host-disease after allogeneic stem cell transplantation. Front Immunol 11:575492. https://doi.org/10.3389/fimmu.2020.575492
Koochakzadeh L, FarrokhpourKarimzadeh H, HaghiAshtiani MT, Safavi M (2020) Trisomy 19 in acute megakaryocytic leukemia. Indian J Hematol Blood Transfusion 36(1):222–224. https://doi.org/10.1007/s12288-019-01154-1
Nimer SD, Macgrogan D, Jhanwar S, Alvarez S (2002) Chromosome 19 abnormalities are commonly seen in aml, m7. Blood 100(10):3838–3839. https://doi.org/10.1182/blood-2002-07-2225
Tadenuma T, Kashiwazaki S (1975) Extra-articular manifestations of rheumatoid arthritis. (1). Splenomegaly. Ryumachi 15(2):122–7 (Japanese)
Zhang M, Zhou L, Xu Y, Yang M, Xu Y, Komaniecki GP, Kosciuk T, Chen X, Lu X, Zou X, Linder ME, Lin H (2020) A stat3 palmitoylation cycle promotes th17 differentiation and colitis. Nature 586(7829):434–439. https://doi.org/10.1038/s41586-020-2799-2
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550
American cancer society (2022) What is rhabdomyosarcoma?. https://www.cancer.org/cancer/types/rhabdomyosarcoma/about/what-is-rhabdomyosarcoma.html. Accessed 19 Oct 2022
Snijders AM, Schmidt BL, Fridlyand J, Dekker N, Pinkel D, Jordan RC, Albertson DG (2005) Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene 24:4232–4242. https://doi.org/10.1038/sj.onc.1208601
Shen X, Zhang H, Xie H, Chen L, Li S, Zheng J, Chai R, Wang Z, Zang Y, He S (2021) Reduced ccr6+il-17a+treg cells in blood and ccr6-dependent accumulation of il-17a+treg cells in lungs of patients with allergic asthma. Front Immunol 12:710750. https://doi.org/10.3389/fimmu.2021.710750
Song X, Tang W, Peng H, Qi X, Li J (2021) Fgfr leads to sustained activation of stat3 to mediate resistance to egfr-tkis treatment. Investig New Drugs 39(5):1201–1212. https://doi.org/10.1007/s10637-021-01061-1
Acknowledgements
We are thankful to those at all sites who contributed to this study in patient recruitment, saliva sample collection, and clinical data collection. We are also thankful to the Oral Medicine Translational Research lab staff for sample processing and storage. This work was supported by funds from the Atrium Health Foundation Research fund.
Funding
This study was supported by the Atrium Health Foundation Research fund.
Author information
Authors and Affiliations
Contributions
Conceptualization: MTB, IVB, BH, AJH, FBM, JLM, KGL.
Manuscript preparation, writing, and revisions: MFB, FBM, JLM, MTB, JEJ.
Data analysis: JLM, MFB, FBM.
All authors read and approved the final submitted version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by Atrium Health IRB# IRB00080071. All participants have signed an informed consent form.
Consent for publication
All authors have approved the final version of the manuscript.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mougeot, JL.C., Beckman, M.F., Hovan, A.J. et al. Identification of single nucleotide polymorphisms (SNPs) associated with chronic graft-versus-host disease in patients undergoing allogeneic hematopoietic cell transplantation. Support Care Cancer 31, 587 (2023). https://doi.org/10.1007/s00520-023-08044-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00520-023-08044-3