
Abstract
Key message
Dehydration and cryo-stress (−196 °C) caused a greater increase in the DNA methylation of embryonic axes (EAs) of beech seeds (tolerant to both stresses) than in EAs of oak (sensitive to both stresses). Simultaneously, in vitro survival of cryopreserved beech EAs was also significantly higher than in those of oak.
Abstract
DNA methylation plays a key role in the regulation of growth and differentiation in plants. The present study focused on global DNA methylation of embryonic axes (EAs) isolated from the seeds of pedunculate oak (Quercus robur L.) and European beech (Fagus sylvatica L.) after cryopreservation. Oak seeds can be labeled recalcitrant, i.e., sensitive to dehydration, while beech seeds fall under the suborthodox category, i.e., tolerant to dehydration. DNA methylation was determined using a specific antibody directed at the methylated cytosine in CpG dinucleotides. The analysis was carried out on DNA isolated from EA tissues and subjected to (1) dehydration (control), (2) a combination of dehydration and plant vitrification solution (PVS3) and (3) dehydration with vitrification PVS3, and rapid cooling in liquid nitrogen (LN2, −196 °C) at a cooling rate of 32 °C s−1. Such treatments induced global DNA methylation primarily in tolerant EAs from F. sylvatica seeds. The level of global DNA methylation in this material was 30 % higher than in the control, i.e., dehydrated (not frozen) beech EAs. The same treatment in oak EAs did not evoke such changes. After 120 days of tissue growth, we observed substantially more DNA demethylation in the beech tissues after cryopreservation in LN2 than in the controls or in tissues subjected to vitrification but not cryopreservation. Information on the global level of DNA methylation can be useful in monitoring procedures aimed at increasing the rate of recovery of EAs following cryopreservation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The European beech (Fagus sylvatica L.) and pedunculate oak (Quercus robur L.) are both important trees for forestry in Europe and are of potential importance in other temperate regions. Both species belong to the family Fagaceae. Oaks and beech are capable of fruiting only after reaching 30–50 years of age. The fruiting is irregular and usually occurs at 4- to 9-year intervals (Suszka et al. 1996). There is, therefore, a need for long-term storage of seeds that are fully viable.
The cryopreservation of embryonic axes is an adequate strategy for long-term storage of Q. robur (Martinez et al. 2003) and many other species (Lambardi et al. 2004; Engelmann 2011; Pukacki and Juszczyk 2015). Q. robur and F. sylvatica seeds are different in respect to their sensitivity to dehydration and freezing stress (Poulsen and Eriksen 1992). Q. robur acorns are characterized by a greater sensitivity to drying, freezing and storage conditions compared to beechnuts. The seeds of F. sylvatica, the storage behavior of which can be categorized as suborthodox (intermediate), tolerate dehydration to water content (WC) of 0.1 g H2O g dm−1, in contrast to the recalcitrant seeds of Q. robur, which show dehydration intolerance to 0.41 g H2O g dm−1 (Pukacka et al. 2003).
The genetic and molecular bases of post-cryo tolerance are poorly understood (Charne et al. 1988; Panis and Lambardi 2006; Folgado et al. 2014). Moreover, little is known about the role of the epigenetic processes involved in the molecular regulation of cold tolerance in embryonic axes.
However, analysis of DNA methylation post-cryo culture can be an indicator of plant adaptation to low temperature stress, as well as regeneration of plant material (Johnston et al. 2009). Evidence suggests that after undergoing stresses, plants may change their response to a future stress (Zeng et al. 2014). This is the theory of ‘stress memory’ (Avramova 2015), according to which information in the primary DNA sequence can be changed as a result of DNA methylation. Moreover, DNA methylation is a hereditary epigenetic phenomenon that plays an important role in the regulation of gene expression, particularly during the growth and development of plants (Bestor and Veredine 2004).
DNA methylation is catalyzed by DNA methyltransferase, which transfers the methyl group from S-adenosylmethionine to cytosine or less often to adenine in the DNA chain to form 5-methylcytosine or 6-methyladenine, respectively. DNA methylation does not modify the DNA sequence (Boyko and Kovalchuk 2011). Epigenetic mechanisms in gene regulation affect plant growth and vary depending on the environment or osmotic stress during a tissue culture (Guevara et al. 2011; Zeng et al. 2014).
Many recent studies have analyzed changes in DNA methylation during in vitro culture of cryopreserved explants (Fernandes et al. 2008; Peredo et al. 2008). The aim of effective cryopreservation is the long-term preservation of plant material, and it is important to know whether this induces permanent genetic changes. Changes in the level of DNA methylation were observed in papaya (Kaity et al. 2008), chrysanthemum (Martín and González-Benito 2006; Martin et al. 2011) currants (Johnston et al. 2009), strawberries (Hao et al. 2002a), citrus (Hao et al. 2002b) and potatoes (Kaczmarczyk et al. 2012). Changes in global DNA methylation may also be induced by cryo-vitrification agents and the conditions of in vitro culturing (Harding 2004). One important question is when and how DNA methylation affects the adaptation of plants to the stress associated with cryostorage and sub-optimal culture conditions in vitro. Epigenetic changes can be affected by all procedures applied in cryopreservation. For example, in cases of cryopreservation applied after vitrification with Me2SO (DMSO) cryoprotectant, reductions in DNA fragmentation were observed (Zilli et al. 2003). Modifications to the chromatin structure and the resulting changes in gene expression are reflected in the structure and function of plants grown in vivo or in vitro. Because DNA methylation is transient, it is crucial to adjust the duration of the analysis to the investigated material (Mikula et al. 2011). Additionally, differences in the intensity of DNA methylation in regenerates stored in LN2 for 3 days versus 24 months have been observed. DNA methylation is an important epigenetic regulation of the expression of genes involved in multiple processes during plant development (Uthup et al. 2013). Significant differences between young and mature meristems of pine (Pinus radiata) have been demonstrated, in which the percentage of DNA methylation in the meristems gradually diminished as their activity increased (Fraga et al. 2002).
The present study expands our knowledge of the impact of changes in DNA methylation status after cryopreservation followed by the post-thaw recovery of embryonic axes of desiccation-tolerant F. sylvatica seeds and desiccation-sensitive Q. robur seeds. We used an immunochemical method to measure the global methylation level in the analyzed probes. This method is safer (non-radioactive), cheaper and less time-consuming than others, but remains very sensitive. Droplet-vitrification is a cryopreservation method that combines vitrification and rapid freezing (Kartha et al. 1982). This knowledge should contribute to the understanding of the epigenetic and molecular processes that occur as a result of cryopreservation and post-thaw culturing in vitro in economically important woody plants.
Materials and methods
Plant material
Mature seeds of F. sylvatica (suborthodox) and Q. robur (recalcitrant) were collected under the canopy of individual trees from populations growing in two forested areas in Hajda (53°23′ N, 16°45′ E) and Płytnica (53°18′ N, 16°47′ E) in north-western Poland. They were transported by car to a laboratory in foil bags in a cooling box in which seeds of Q. robur were dried to a water content (WC) of 0.82 g H2O g dm−1; F. sylvatica seeds were dried to WC 0.33 g H2O g dm−1. The seeds were then stored at −3 °C in polythene bags until use.
Stratification of beech seeds
Prior to the experiments, the beech seeds required cold stratification to exit from dormancy. The seeds of Q. robur were not dormant. The set of fully imbibed F. sylvatica seeds was stratified in 7.0 × 18.0 cm plastic boxes containing a 2-cm layer of moist rolled paper at 3 °C for 8–11 weeks in phytotron (Suszka et al. 1996). A seed was scored as germinated when the radicle protruded 2 mm. Germination counts were made every week, and the germinated seeds were removed (ISTA 1999). When the first seeds had germinated, the stratification process was finished. These stratified beech seeds were used for all experiments.
Dehydration of EAs and water content determination
Cleaned and surface-sterilized seeds of F. sylvatica and Q. robur EAs were separated from their cotyledons and placed on a piece of filter paper that was moistened with sterile distilled water in a closed Petri dish prior to the dehydration process. Then, the EAs were dehydrated in an apparatus used by Wesley-Smith et al. (2001a) with some modifications. They were placed on nylon gauze inserted across a PVC pipe (95 mm diameter, 220 mm in length), which acted as a support. A computer CPU cooling fan (12 V, 1 W, 90 mm diameter) was mounted in the middle of this PVC pipe with airflow directed toward the gauze at 17 % relative humidity (RH). Activated silica gel (500 g) was placed at the bottom of the jar. The dehydration was applied for different durations (10–240 min). A digital thermo/hygrometer was used to monitor the temperature and RH in the apparatus during dehydration (Pukacki and Juszczyk 2015).
The water content of the EAs was determined gravimetrically (105 °C for 48 h). Three samples of 20 axes in three replicates were used for each determination. The water content was expressed as g H2O (g dry mass)−1.
Differential thermal analysis (DTA) of cellular water
DTA was used to determine the ice nucleation of supercooled tissue water. Single embryonic axes were wrapped in aluminum foil and placed in the DTA instrument (Pukacki and McKersie 1990; Pukacki et al. 1991). The four samples were analyzed simultaneously. This technique was used to determine the difference in temperature between the tissues and a reference dry sample. The samples were cooled from 20 to −40 °C and rewarmed to 20 °C at 0.11 °C min−1 (Pukacki and Juszczyk 2015). In these experiments, copper-constantan thermocouples (0.1 mm in diameter) were held to the surface of the axes by wrapping them together with laboratory film (Parafilm, American Can Corp., Greenwich, CT, USA). The course of the analysis was controlled and recorded with the aid of an IBM computer equipped with an analog/digital board (Type WB-AAI-B8) and software developed by Omega Engineering, Stamford, USA. Onset temperatures for ice nucleation in single embryonic axes were calculated from the point of discontinuity in the baseline. Exothermic data were collected from eight replicates per treatment. After thermal analysis, individual samples were dried at 103 °C for 24 h, and their dry mass was determined.
Droplet-vitrification and cryopreservation of EAs
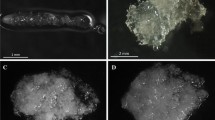
The embryonic axes (after dehydration as described above) were submitted to vitrification in cold PVS3 solution containing 50 % (w/v) sucrose and 50 % (w/v) glycerol (Nishizawa et al. 1993) or with PVS2 containing 30 % glycerol, 15 % ethylene glycol and 15 % DMSO in 0.4 M sucrose in liquid medium (Sakai et al. 1990) w/v for 60 min at room temperature. Adding chilled solutions on ice may reduce the toxic effects of vitrification. Ten samples of 5 EAs of each species were dehydrated to various water contents. After pre-dehydration, the samples were separately placed onto a sterile strip of aluminum foil (60 mm long × 5 mm wide × 0.05 mm deep) with 25 µl droplets of cold PVS3 or PVS2 solution and then plunged immediately into liquid nitrogen, in which they were cooled at 32 °C s−1 (from 0 to −150 °C) for 2 h (Fig. 1). After LN2 exposure, the Al strips were rapidly thawed and transferred to a thermostat containing a liquid culture of half-strength Woody Plant Medium (WPM) (Lloyd and Mc Cown 1980) at 20 ± 1 °C for 30 min. The embryonic axes were warmed at a rate of 28 °C s−1 to 0 °C, washed in water three times, and then stored in 0.1 M sucrose at 0 °C for 20 h. The EAs were then surface-sterilized (Wesley-Smith et al. 2001b) using a 3-step protocol: (1) 0.1 % mercuric chloride for 10 min, (2) 10 % “Domestos” (sodium hypochlorite NaClO <5 %) for 10 min, and (3) 75 % ethyl alcohol for 1.0 min and rinsed in sterile water. They were then transferred for recovery.

Pedunculate oak or beech embryonic axes placed in drops of PVS3 on aluminum foil strips prior to being plunged in liquid nitrogen (−196 °C). White sticks indicate of embryonic axes in vitrification solution
In vitro culture of EAs
The embryonic axes were cultured on Woody Plant Medium (WPM) solidified with 0.7 % (w/v) agar. The pH of the medium was adjusted to 5.5 before sterilization. The Petri dishes containing the recovering axes were stored for 6 days in the dark and then maintained under a 16:8 h light/dark photoperiod with illumination from cool white fluorescent light (40 µmol m−2 s−1), in a growth chamber under standard conditions (23 ± 1 °C), (Mytron WB 750 KHFL Germany). The surviving axes were scored as ‘normal’ if the radicle developed and shoots expanded and turned green (Wesley-Smith et al. 2014). After 3 weeks, surviving EAs were transferred onto the same WPM medium in culture tubes (150 × 25 mm). The survival of seedlings from the EAs were recorded after 6 weeks of culture as a percentage of the total number of EAs. The seedlings were scored as survived when growth was observed in both the roots and the shoots (Pukacki and Juszczyk 2015).
DNA methylation analysis
Embryonic axes subjected to different treatments (described above) were cultured in vitro on WPM medium. The determination of global DNA methylation was performed on DNA isolated from 25 mg of cultured tissues collected after 20, 30 and 120 days of culture, respectively. DNA was isolated using a DNeasy Plant Mini Kit (Promega), (Gryzinska et al. 2013). This procedure is based on the interaction of nucleic acids with a silica-based membrane after cell lysis and RNA digestion, using RNase without phenol extraction or ethanol precipitation. This procedure enables the acquisition of good quality DNA without RNA contamination. Analysis of the global level of DNA methylation in F. sylvatica and Q. robur tissues was performed using an Imprint Methylated DNA Quantification kit (Sigma-Aldrich). Methylated DNA was detected using antibodies specific to methylated cytosine in the CpG dinucleotide. We used 200 ng of DNA (each well can bind a maximum of 200 ng total DNA). DNA binding was performed during the incubation of DNA at 37 °C for 1 h; a blocking solution was then added and the samples were incubated again at 37 °C for 30 min. Methylated DNA was then captured using a specific antibody and detected by binding to a detection antibody (Oakeley et al. 1997). After the addition of a developing solution to the wells, a colorimetric reaction occurred (the solution turned blue). These complexes were then colorimetrically quantified by measuring absorbance at a wavelength of 450 nm. Both the blank control and methylated control (positive control) were analyzed together with the DNA samples. Using this method, we can determine the relative level of global DNA methylation. A standard curve was prepared using control methylated DNA (provided with the kit). The amount of methylated DNA present in the sample was proportional to the absorbance measured (Vassar et al. 2009). The advantage of this method is its reproducibility, sensitivity and short duration compared to other methods. Using this method one can calculate the methylation level in the probe relative to the methylated control DNA. The analyses were performed in 3 replicates. The replicates were averaged, and the mean value was used for further analyses. The following formula was used to calculate the amount of methylated DNA in the total DNA [%]:
where OD is the optical density; M3 is a blank probe; S is the amount of input sample DNA in ng; M4 is the positive control, a methylated polynucleotide containing 50 % of 5-methylcytosine; and P is the amount of input-positive control in ng.
The amount of methylated DNA was expressed as a percentage of global DNA.
Statistical analysis
The data are presented as the mean ± SD. The data for all seedlings throughout the treatment process were subjected to an analysis of variance (ANOVA) using Statgraphics Plus software. Significant differences between the treatments were compared using the Newman-Keuls range test, and significance was determined when P < 0.05. For all regression analyses, the percentages were transformed (arcsin) such that the data conformed to parametric test assumptions.
Results
Differential thermal analysis (DTA) of tissue water
Desiccated EAs from seeds of both species were subjected to DTA determination to determine how the ice nucleation temperature is affected by water content (Fig. 2). On the cooling scans, the exothermic peak corresponded to the crystallization of supercooled water (Fig. 2a). The temperature of ice nucleation in EAs decreased with decreasing WC in this material. The temperature of the freezing transition decreased from approximately −7 °C in the axes of both species to approximately −27 °C for recalcitrant Q. robur after dehydration to WC 0.55 g H2O g dm−1 and to −21 °C for suborthodox F. sylvatica after dehydration to WC 0.35 g H2O g dm−1 of embryonic axes (Fig. 2b). DTA showed that ice nucleation was not observed below a certain threshold of WC. In axes of F. sylvatica, the WC threshold was 0.33 g H2O g dm−1; for the Q. robur axes, this threshold was 0.54 g H2O g dm−1. The ice nucleation temperatures of water at these thresholds were −21.0 and −26.0 °C, respectively. A significant (P < 0.001) positive correlation was found between WC and the exothermic onset of ice nucleation for both EAs [F. sylvatica (r 2 = 0.681) and Q. robur (r 2 = 0.861)] (Fig. 2b). Based on the aforementioned results, the selected levels of EAs dehydration for cryopreservation were 0.25 and 0.43 g H2O g dm−1 for F. sylvatica and Q. robur, respectively. These conditions induced limited injury of EAs due to ice nucleation during the freezing process and good regeneration for plants grown in vitro.

Representative DTA scans a during the cooling of beech single embryonic axes after dehydration to different moisture levels. This indicated that the onset of ice nucleation occurred at −7.6 °C at WC 2.12 g H2O g dm−1 and at 21.1 °C at WC 0.43 g H2O g dm−1. b Relationship between water content and ice nucleation temperatures during cooling in single embryonic axes of F. sylvatica and Q. robur (P < 0.001)
Survival following dehydration, vitrification and cryopreservation of EAs
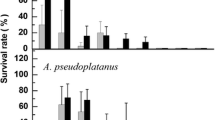
The embryo axes of both tree species (F. sylvatica and Q. robur) were dehydrated to WC 0.25 or 43 g H2O g dm−1, respectively, subjected to vitrification for 60 min in PVS2 or PVS3, and then submerged in LN2 to retain 70 and 40 % post-cryo survival, respectively (Fig. 3). Exposure to dehydration and vitrification in PVS2 for 60 min followed by cryopreservation in LN2 resulted in a decrease in post-thaw axes regrowth in F. sylvatica to 43 % and Q. robur to 30 % compared with vitrification in PVS3 (Fig. 3). In the controls, the survival of dehydrated EAs slowly decreased as the water content of these untreated axes was reduced, which led to a significant reduction in seedling recovery (Fig. 4). The fully hydrated control EAs of both species that had not been exposed to LN2 and had not undergone vitrification in the presence of PVS3 germinated at 80 and 100 %, respectively (Fig. 4). The rate of survival and development of EAs in suborthodox F. sylvatica remained high (70 %) when the WC was reduced to 0.25 g H2O g dm−1 (r 2 = 0.859) (Table 1; Fig. 4). Dehydration to WC 0.66 g H2O g dm−1 caused a 77 % reduction in the survival rate (Table 1; Fig. 4). For Q. robur, dehydration to a low WC level of 0.25 g H2O g dm−1 evoked a significant reduction in the survival rate of embryonic axes to 9 % (r 2 = 0.941). However, dehydration to WC 0.43 g H2O g dm−1 and preculture in PVS3 solution was insufficient to guarantee the viability of Q. robur EAs. The positive effect of the combination of these two treatments (dehydration and the application of PVS3-based vitrification) is unquestionable (Fig. 5). Recovery, corresponding to the production of normal seedlings from treated EAs, was observed after 30 days (Fig. 5). Dehydration and vitrification in PVS3 solution followed by cryopreservation in LN2 resulted in a loss of viability in F. sylvatica and in Q. robur, when the WCs were 1.0 and 0.66 g H2O g dm−1, respectively. This occurred due to intracellular ice in the embryonic axes. The highest (over 40 %) survival for suborthodox F. sylvatica EAs appeared when they had been dehydrated to WC 0.25 g H2O g dm−1 and subjected to cryopreservation in LN2. However, for Q. robur, the highest survival score (35 %) was recorded with a combination of dehydrating the axes to WC 0.43 g H2O g dm−1 plus PVS3 and LN2 treatments (Fig. 4). A linear regression revealed highly significant (P < 0.01) positive correlations between WC and the survival rate of EAs subjected to vitrification and cryopreservation (r 2 = 0.882 for F. sylvatica and r 2 = 0.732 for Q. robur) (Table 1).

Effect of different pretreatment times with PVS2 or PVS3 vitrification solutions on survival of F. sylvatica and Q. robur embryonic axes following exposure to LN2. Embryonic axes of F. sylvatica and Q robur before treatments were dehydrated to WC 0.25 H2O g dm−1and WC 0.43 H2O g dm−1, respectively. Error bars represent ± SD of means of 5 replications. Asterisks indicate values that are significantly different from the control according to the Newman-Keuls range test; (P < 0.05)

Relationship between water content and survival of F. sylvatica and Q. robur embryonic axes during the steps of cryopreservation: dehydration to WC 0.25 H2O g dm−1and WC 0.43 H2O g dm−1, respectively, dehydration (control, non cryo-stored samples), dehydration-vitrification in PVS3 and after both cycles; dehydration-vitrification and exposure to LN2. Assessments were performed for up to 6 months after culture of embryonic axes on WPM medium. The results are expressed as the mean ± SD of ten embryonic axes with three replicates. Asterisks indicate values that are significantly different from the control according to the Newman-Keuls range test; (P < 0.05)

In vitro regeneration of seedlings from embryonic axes after dehydration, pretreatment with PVS3 and cryogenic exposure in LN2, of F. sylvatica (a–c), and Q. robur (e–g), after 20, 30, 120 days of culture on WPM medium. Plants of F. sylvatica (d) and Q. robur (h) originated from cryopreserved embryonic axes, regenerated plants growing in the growth chamber
DNA methylation during in vitro culture
In fully hydrated (control) axes of F. sylvatica growing at standard conditions, the global DNA methylation levels estimated at 20, 30, and 120 days were 36.8, 16.7 and 21.4 %, respectively (Fig. 6). The highest methylation was obtained after 30 days of F. sylvatica embryonic axes growth following either vitrification or vitrification following cryopreservation. In EAs precultured in vitrification PVS3 solution or following cryopreservation, the global methylation level on the 30th day increased to 44.6 % from approximately 3.0 % observed on the 20th day of cultivation. However, on the 120th day of growth, the global methylation decreased to 13.7 %, which was the lowest value in the F. sylvatica samples.

Effects of cryopreservation steps on F. sylvatica and Q. robur embryonic axes after dehydration only (control); dehydration and pretreatment with PVS3; and both cycles and exposure in LN2, on global DNA methylation during seedling development from embryonic axes on WPM medium. The hypocotyls of seedlings were collected after 20, 30 and 120 days of culture on regeneration medium of embryonic axes. Data are mean ± SD (n = 5). Asterisks indicate values that are significantly different from the control (non cryo-stored samples) at the time, according to the Newman-Keuls range test; (P < 0.05)
In the case of Q. robur, the DNA methylation after the above-mentioned treatments and during in vitro culture led to substantially fewer changes. No significant differences were recorded during seedling development between the treated samples and the controls. The degrees of global DNA methylation in the control samples of Q. robur during the growth periods of 20, 30 and 120 days were 22.4, 16.9 and 12.8 %, respectively (Fig. 6). A significantly higher global DNA methylation level (23.1 %) was observed in PVS3-treated EAs after 30 days of growing in vitro (Fig. 6). This suggests that vitrification enhanced DNA methylation during the first 4 weeks of growth. After 17 weeks, there was demethylation in all axes of both species regardless of the treatment. The lowest methylation levels (13.7 and 8.1 %) were reached in F. sylvatica and Q. robur EAs, respectively, after 120 days of growth in the cryopreserved axes (Fig. 6).
Discussion
Thermal analysis of cellular water
The successful cryopreservation of EAs can be ensured by controlling or avoiding intracellular ice nucleation. Ice nucleation in living cells evokes serious consequences. When liquid water is removed from the cells to the extracellular space, solutions within the cell become more concentrated and heterogeneous ice nucleation is reduced. During this process, an important role is played by the level of tissue hydration and the presence or absence of ice nucleators (Jarząbek et al. 2009; Wisniewski et al. 2009; Ishikawa 2014). DTA was used to determine the ice nucleation of supercooled tissue water. Selected freezing thermograms of dehydration-treated axes dried to various moisture contents are presented in the Fig. 2a. The DTA scan shows that water is supercooled during the cooling of EAs. This is followed by an exothermic peak reflecting ice crystallization (Fig. 2). In the extreme dehydration of tissues, most or all freezable water is removed from the cells and vitrification of the internal solution occurs during rapid cooling. This avoids lethal intracellular ice crystallization (Engelmann 2008). The DTA results showed that ice nucleation was not observed below a certain WC threshold (Fig. 2b).
Recovery of embryonic axes
Seeds of species belonging to the suborthodox category can be safely dehydrated to lower moisture levels than species that are recalcitrant; thus, the cryopreservation of suborthodox EAs has a greater chance of success despite release from dormancy (Pukacka and Ratajczak 2014). To date, successful freezing in LN2 has been reported for EAs in recalcitrant seeds of Acer pseudoplatanus (Pukacki and Juszczyk 2015), A. saccharinum (Beardmore and Whittle 2005), Quercus suber and Q. ilex (González-Benito et al. 2002). Seeds from the suborthodox category are intermediate between the orthodox and recalcitrant categories and show a high sensitivity to low temperatures between 0 and −20 °C (Hong and Ellis 1990). Suborthodox seeds fall from the trees highly hydrated because they are not subject to natural dehydration (Berjak and Pammenter 2008). These seeds are tolerant to dehydration to WC 0.11 g H2O g dm−1 (similar to orthodox seeds), and cryopreservation of their EAs is substantially more effective (Poulsen 1992). During extreme dehydration of explants, most or all freezable water is removed from cells, and vitrification of the internal solutes takes place during rapid exposure to LN2 (Engelmann 2008). Notably, the seeds of the studied species differ in their ability to germinate after being harvested from trees. The F. sylvatica seeds are characterized by deep dormancy, whereas those of Q. robur are dormant for only a short period after seed maturation. The dormancy of F. sylvatica seeds can be broken by cold stratification at 3 °C (Suszka et al. 1996). In this study, Q. robur and F. sylvatica EAs were dehydrated to WC 0.43 and 0.25 g H2O g dm−1, respectively, and then subjected to vitrification in PVS3 solution, thereby enabling safe freezing in liquid nitrogen and post-thaw in vitro regeneration. We found that 40 and 35 % of EAs of F. sylvatica and Q. robur, respectively, survived freezing in LN2. Successful cryopreservation was obtained for Quercus nigra and Q. falcata, the EAs of which were dehydrated to WC 0.25 or 0.43 g H2O g dm−1 (Pence 1990). A negative impact on the survival of the EAs was noted when they were treated with PVS2 vitrification solution (Fig. 3 ). Some studies have implied a relationship between increased survival and higher DMSO content (Reed and Uchendu 2008). The present study showed that the dehydration of EAs of F. sylvatica to WC 0.66 g H2O g dm−1 resulted in 90 % survival, and that further dehydration to 0.25 g H2O g dm−1 reduced survival to 70 %, indicating that F. sylvatica EAs were tolerant to dehydration to a low WC (Fig. 4). Another study found that the dehydration of whole seeds to WC 0.25 g H2O g dm−1 in Q. robur reduced survival to 25 %; however, 93 % of the excised EAs survived (Finch-Savage 1992). In our study, the survival of EAs of both species was high following vitrification in PVS3 and dehydration to WC 0.43 and 0.66 g H2O g dm−1. Exposure to PVS3 osmotically dehydrates cells at a non-freezing temperature, and a vitrification solution subjects cells to osmotic stress, thereby making it very likely that some constituents will enter the cells. This increases cytoplasm viscosity and decreases the glass transition temperature. However, the penetrative nature of some cryoprotectants may also result in toxicity (Fig. 3). A significant decrease in survival occurred after EAs were exposed to LN2. The freezing of EAs of both species in LN2 at a high WC (1.0 g H2O g dm−1) led to cellular damage via intracellular ice formation. This was a consequence of damage to cytoplasmic membranes, which results in increased ion flux (Pukacki and Juszczyk 2015). Reactive oxygen species (ROS), such as superoxide anion (O ·−2 ), hydrogen peroxide (H2O2) and hydroxyl radical (·OH), represent direct factors contributing to membrane injury and reduced EAs viability during the stages of cryopreservation (dehydration-vitrification and then plunging in LN2) (Juszczyk and Pukacki 2013; Pukacki et al. 2011). Similar evidence has been shown in studies of freezing stress in wheat meristems (Kendall and McKersie 1989) and Arabidopsis seedlings (Chen et al. 2015). ROS can alter the physico-chemical properties of membranes, causing them to lose a liquid-crystalline structure (McKersie and Leshem 1994). The excessive production of ROS can affect DNA sequences, gene expression and protein profiles, which in turn may lead to mutational changes and cell death (Olinski et al. 2003; Harding 2004; Demidchik 2015; Ito and Kuraoka 2015). Therefore, an efficient system utilizing antioxidants (low-molecular-weight antioxidants and enzymes) during the post-cryo recovery period in the dark (6 days) could prevent or reduce the associated detrimental effects on frozen samples (Pukacki and Juszczyk 2015), and thus facilitate survival and development (Uchendu et al. 2010; Chen et al. 2015).
DNA methylation during in vitro culture
Symmetric methylation occurs most frequently (within CpG dinucleotide sites), whereas asymmetric methylation occurs less frequently (at CpNpNp sites). Studies of various organisms have shown that DNA methylation is correlated with the methylation of lysine 9 of histone H3 (H3K9) (Wierzbicki and Jerzmanowski 2005). DNA methylation is involved in many important biological of processes, such as the regulation of gene expression, gene silencing and genomic stigma (Guevara et al. 2011), and plays an important role in plant phenology (Wisniewski et al. 2015). There are many methods for determining DNA methylation in biological material. In this paper, we determined global DNA methylation using antibodies directed against methylated cytosine in the CpG dinucleotide. The highest level of global DNA methylation in samples subjected to cryopreservation compared to a control was observed after 30 days of culture. This high level of methylation was not maintained for an extended duration and was significantly lower after 120 days of seedling culture (nearly equal to the control values) (Fig. 6). The more resistant species of F. sylvatica had a stronger response than the dehydration-sensitive Q. robur; however, in F. sylvatica, methylation occurred during the same stages of the protocol. Such changes were not observed in the recovered plants after the culture cycle. The long-term implications of these temporal changes on the genetic stability of cryopreserved shoots are currently unknown. These findings suggest that molecular changes may be essential in modulating the tolerance of F. sylvatica tissues to cold stress. Similarly, the levels of DNA methylation were decreased in the meristems of currants (Ribes) with susceptible genotypes; in tolerant genotypes, the number of methylated DNA sequences increased compared to controls (Johnston et al. 2009). However, research by Peredo et al. (2009) did not show epigenetic changes in two hop accessions (Humulus lupulus) after 2 years of in vitro micropropagation and after 12 cycles compared to field plants. In the present study, the levels of DNA methylation were high in both F. sylvatica (36.9 %) and Q. robur (22.4 %) in control tissues cultured for 20 days. The difference in global DNA methylation between F. sylvatica and Q. robur was significant. This result can be interpreted as a genetic property, and these differences may be due to the physiologically higher growth rate of EAs of F. sylvatica seedlings compared to Q. robur seedlings. However, after 30 days of post-thaw EAs culture, global DNA methylation remained highest in F. sylvatica tissues treated with PVS3 solution only; this was also observed in those cryopreserved in LN2 (44.6 and 47.8 %, respectively). In Q. robur, the corresponding values were 23.2 and 16.6 %. A further post-cryo culture of seedlings (to 120 days) caused a significant decrease in DNA methylation in both species compared to the control (to 13.7 % for F. sylvatica and 8 % for Q. robur) (Fig. 6). This is consistent with differences in global DNA methylation between meristematic areas of juvenile and mature Pinus radiata (Fraga et al. 2002). In contrast, no changes in DNA methylation levels were observed in regenerated shoots of Ribes during 140 days of in vitro culture (Johnston et al. 2009). These data indicate that global DNA methylation in the seedlings that developed from EAs after cryopreservation was not static and that it had unique dynamics during each developmental stage.
To achieve more effective cryopreservation via vitrification in this work, we used PVS3 vitrification solution instead of the more dangerous PVS2 solution. However, a positive effect for PVS2 solution on tissue vitrification has also been reported (Chartest et al. 1996; Maślanka et al. 2013). In our experiments, vitrification with PVS2 solution, which contains toxic DMSO, significantly reduced the survival of EAs (depending on the duration of treatment) compared to vitrification in PVS3 (Fig. 3). Kaity et al. (2008) studied the genetic stability of papaya shoots during cryopreservation after vitrification with PVS2. Those authors showed that the observed changes could be related to the toxicity of DMSO as a component of PVS2. DNA methylation plays a key role in the control of plant development (Perez et al. 2015); thus, in our study, the expression of genes responsible for the rooting of seedlings may have started after 30 days of post-thaw culture. The stronger development of the roots of F. sylvatica was likely a result of major changes in DNA methylation in hypocotyls. The genetic stability of cryopreserved plant explants is usually high. Differences between the control tissues and frozen tissues were not explained by the influence of the cryopreservation process, but were thought to result from tissue regeneration following cryopreservation (Harding 2004).
In our study, demethylation was detected in seedlings of both species after 120 days. Demethylation has not been thoroughly studied. This process (in the case of cryopreserved EAs) was nearly fourfold more intensive in F. sylvatica than in Q. robur and amounted to 34 and 8 %, respectively. Similarly, remarkable demethylation has been found in cryopreserved explants of cultivars of Humulus lupulus (Peredo et al. 2008). Substantial demethylation of DNA during cryopreservation has also been demonstrated in Fragaria gracilis (Hao et al. 2002a) and Malus pumila (Hao et al. 2002b). To a lesser extent, but with a similar trend, global DNA demethylation occurred in EAs subjected only to vitrification in PVS3. These results are consistent with the hypothesis of Viejo et al. (2012) stating that demethylation is necessary for changes in gene expression to initiate each stage of ontogenetic development in plants.
Conclusions
The EAs of F. sylvatica are characterized by a significantly better growth post-cryo survival rate and higher tolerance to dehydration and low temperatures than the EAs of Q. robur. The best conditions for post-cryo survival of EAs of both species was dehydration to WC 0.25 and 0.43 g H2O g dm−1 for F. sylvatica and Q. robur, respectively, and vitrification with PVS3 for 60 min prior to cryopreservation. This may be linked with the greater tolerance to desiccation of F. sylvatica embryonic axes. However, the long-term implications of these temporal changes on the genetic stability of cryopreserved embryonic axes have not yet been determined, so further analyses are needed to confirm whether the stress caused by cryopreservation influences the behavior of plants in field conditions, as well.
Author contribution statement
PM. P., conceived and designed the experiments, and supervised the biophysical analysis contributed to data interpretation and prepared the manuscript. K. N., analyzed DNA methylation and interpreted these data with assistance from M. M. Seed collection and in vitro culture of embryonic axes were carried out by PM. P.
References
Avramova Z (2015) Transcriptional ‘memory’ of a stress: transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J 83:149–159
Beardmore T, Whittle CA (2005) Induction of tolerance to desiccation and cryopreservation in silver maple (Acer saccharinum) embryonic axes. Tree Physiol 25:965–972
Berjak P, Pammenter NW (2008) From Avicennia to Zizania: seed recalcitrance in perspective. Ann Bot 101:213–228
Bestor TH, Veredine GL (2004) DNA metylotransferases. Curr Opin Cell Biol 6:380–389
Boyko A, Kovalchuk I (2011) Genome instability and epigenetic modification-heritable responses to environmental stress? Opin Curr Plant Biol 14:260–266
Charne D, Pukacki P, Kot L, Beversdorf WD (1988) Embryogenesis following cryopreservation in isolated microspores of rapeseed (Brassica napus L.). Plant Cell Rep 7:407–409
Chartest PJ, Bonga J, Klimaszewska K (1996) Cryopreservation of plant tissue cultures: the example of embryogenic tissue cultures from conifers. Plant Tissue Cult Man C9:1–27
Chen G, Ren L, Zhang J, Reed BM, Di Z, Shen X (2015) Cryopreservation affects ROS-induced oxidative stress and antioxidant response in Arabidopsis seedlings. Cryobiology 70:38–47
Demidchik V (2015) Mechanisms of oxidative stress in plants: from classical chemistry to cell biology. Environ Exp Bot 109:212–228
Engelmann F (2008) The development of eucapsulation dehydration. In: Reed BM (ed) Plant cryopreservation: a practical guide. Springer Sci-Business Media, New York, pp 59–75
Engelmann F (2011) Cryopreservation of embryos: an overview. In: Thorpe AT, Yeung EC (eds) Plant embryo culture: methods and protocols, methods in molecular biology, vol 710. Springer Sci-Business Media, New York, pp 155–183
Fernandes P, Rodriguez E, Pinto G, Roldan-Ruiz I, De Loose M, Santos C (2008) Cryopreservation of Quercus suber somatic embryos by encapsulation-dehydration and evaluation of genetic stability. Tree Physiol 28:1841–1850
Finch-Savage WE (1992) Embryo water status and survival in the recalcitrant species Quercus robur L: evidence for a critical moisture content. J Exp Bot 43:663–669
Folgado R, Sergeant K, Renaut J, Swennen R, Hausman J-F, Panis B (2014) Changes in sugar content and proteome of potato in response to cold and dehydration stress and their implications for cryopreservation. J Proteomics 98:99–111
Fraga MF, Rodriguez R, Canal MJ (2002) Genomic DNA methylation–demethylation during aging and reinvigoration of Pinus radiata. Tree Physiol 22:813–816
González-Benito ME, Garcia-Martin G, Manzanera JA (2002) Shoot development in Quercus suber L. somatic embryos. In Vitro Cell Dev Biol Plant 38:477–480
Gryzinska M, Blaszczak E, Strachecka A, Jezewska-Witkowska G (2013) Analysis of age-related global DNA methylation in chicken. Biochem Genet 51:554–563
Guevara A, Sáez E, Díaz L-M, Herranz T, Barbero C, Vélez D, Collada C (2011) DNA methylation and adaptive response in forest tree species. BMC Proceedings 5(Supl 7):P86
Hao YJ, You CX, Deng XX (2002a) Analysis of ploidy and patterns of amplified fragment polymorphism and methylation sensitive amplified polymorphism in strawberry plant recovered from cryopreservation. CryoLetters 23:37–46
Hao YJ, You CX, Deng XX (2002b) Effects of cryopreservation on developmental competency, cytological and molecular stability of Citrus callus. CryoLetters 23:27–35
Harding K (2004) Genetic integrity of cryopreserved plant cells: a review. CryoLetters 25:3–22
Hong TD, Ellis RH (1990) A comparison of maturation drying, germination, and desiccation tolerance between developing seeds of Acer pseudoplatanus L. and Acer platanoides L. New Phytol 116:589–596
Ishikawa M (2014) Ice nucleation activity in plant tissues. Cryobiol Cryotechn 60:79–88
ISTA (1999) International rules for seed testing. Rules 1999. Seed Sci Technol 27:1–133 (Supplement)
Ito S, Kuraoka I (2015) Epigenetic modifications in DNA could mimic oxidative DNA damage: a double-edged sword. DNA Repair 32:52–57
Jarząbek M, Pukacki PM, Nuc K (2009) Cold-regulated proteins with potent antifreeze and cryoprotective activities in spruces (Picea spp.). Cryobiology 58:268–274
Johnston JW, Benson EE, Harding K (2009) Cryopreservation induces temporal DNA methylation epigenetic changes and differential transcriptional activity in Ribes germplasm. Plant Physiol Biochem 47:123–131
Juszczyk K, Pukacki PM (2013) Threats during cryopreservation of seed embryonic axes of woody plants. Sylwan 157:842–853 (in Polish)
Kaczmarczyk A, Funnekotter B, Menon A, Phang PY, Al-Hanbali A, Bunn E, Mancera RL (2012) Current issues in plant cryopreservation, In: Katkov II (ed), Current Frontiers in Cryobiology. In Tech, Croatia, pp 417–438
Kaity A, Ashmore SE, Drew RA, Dulloo ME (2008) Assessment of genetic and epigenetic changes following cryopreservation in papaya. Plant Cell Rep 27:1529–1539
Kartha KK, Leung NL, Mroginski LA (1982) In vitro growth responses and plant regeneration from cryopreserved meristems of Cassava (Manihot esculenta Crantz). Z Pflanzenphysiol 107:133–140
Kendall EJ, McKersie BD (1989) Free radical and freezing injury to cell membranes of winter wheat. Physiol Plant 76:86–94
Lambardi M, De Carlo A, Biricolti S, Puglia AM, Lombardo G, Siragusa M, De Pasquale F (2004) Zygotic and nucellar embryo survival following dehydration/cryopreservation of Citrus intact seeds. CryoLetters 25:81–90
Lloyd G, Mc Cown B (1980) Commercially-feasible micropropagation of mountain Laurel, Kalmia latifolia, by use of shoot tip culture. Pro Intr Prop Soc 30:421–427
Martin C, Cerverab MT, González-Benito ME (2011) Genetic stability analysis of chrysanthemum (Chrysanthemum × morifolium Ramat) after different stages of an encapsulation–dehydration cryopreservation protocol. J Plant Physiol 168:158–166
Martín C, González-Benito ME (2006) Comparison in somaclonal variant cryopreserved Dendranthema grandiflora shoot apices. Cryobiology 53:367–446
Martinez MT, Ballester A, Vieitez AM (2003) Cryopreservation of embryogenic cultures of Quercus robur using desiccation and vitrification procedures. Cryobiology 46:182–189
Maślanka M, Panis B, Bach A (2013) Cryopreservation of Galanthus elwesii Hook. Apical meristems by droplet-vitrification. CryoLetters 34:1–9
McKersie BD, Leshem YY (1994) Desiccation. In: McKersie BD, Lesham YY (eds) Stress and stress coping in cultivated plants. Kluwer Academic Publishers, Dordrecht, pp 132–147
Mikula A, Tomiczak K, Rybczyński JJ (2011) Cryopreservation enhances embryogenic capacity of Gentiana cruciata (L.) suspension culture and maintains (epi)genetic uniformity of regenerants. Plant Cell Rep 30:565–574
Nishizawa S, Sakai A, Amano Y, Matsuzawa T (1993) Cryopreservation of asparagus (Asparagus officinalis L.) embryogenic suspension cells and subsequent plant regeneration by vitrification. Plant Sci 91:67–73
Oakeley EJ, Podesta A, Jost JP (1997) Developmental changes in DNA methylation of the two tobacco pollen nuclei during maturation. Proc Natl Acad Sci USA 94:11721–11725
Olinski R, Gackowski D, Rozalski R, Foksinski M, Bialkowski K (2003) Oxidative DNA damage in cancer patients: a cause or a consequence of the disease development? Fundam Mol Mech Mutagen 531:177–190
Panis B, Lambardi M (2006) Status of cryopreservation technologies in plants (crops and forest trees). In: Ruane J, Sonnino A (eds) The role of biotechnology in exploring and protecting agricultural genetic resources. Food and Agriculture Organization of the United Nations, Rome, pp 61–78
Pence VC (1990) Cryostorage of embryo axes of several large-seeded temperate tree species. Cryobiology 27:212–218
Peredo EL, Arroyo-García R, Reed BM, Revilla MA (2008) Genetic and epigenetic stability of cryopreserved and cold-stored hops (Humulus lupulus L.). Cryobiology 57:234–241
Peredo EL, Arroyo-García R, Reed BM, Revilla MA (2009) Epigenetic changes detected in micropropagated hop plants. J Plant Physiol 166:1101–1111
Perez M, Viejo M, La Cuesta M, Toorop P, Canal MJ (2015) Epigenetic an hormonal profile during maturation of Quercus suber L. somatic embryos. J Plant Physiol 173:51–61
Poulsen KM (1992) Sensitivity to desiccation and low temperatures (−196°C) of embryo axes from acorns of the pedunculate oak (Quercus robur L.). CryoLetters 13:75–82
Poulsen KM, Eriksen EN (1992) Physiological aspects of recalcitrant in embryonic axes of Quercus robur L. Seed Sci Res 2:215–221
Pukacka S, Ratajczak E (2014) Factors influencing the storability of Fagus sylvatica L. seeds after relase from dormancy. Plant Growth Reg 72:17–27
Pukacka S, Hoffmann SK, Goslar J, Pukacki PM, Wójkiewicz E (2003) Water and lipid relation in beech (Fagus sylvatica L.) seeds and its effect on storage behavior. BBA 1621:48–56
Pukacki PM, Juszczyk K (2015) Desiccation sensitivity and cryopreservation of the embryonic axes of the seeds of two Acer species. Trees 29:385–396
Pukacki PM, McKersie BD (1990) Supercooling and ice nucleation events in the crown of winter wheat seedlings. Can J Plant Sci 70:1179–1182
Pukacki PM, Kendall EJ, McKersie BD (1991) Membrane injury during freezing stress to winter wheat (Triticum aestivum L.) crowns. J Plant Physiol 138:516–521
Pukacki PM, Zenkteler E, Guźniczak E (2011) Cryopreservation of embryonic axes of Fagus sylvatica in relation to dehydration and thermal properties of tissue water. In: Cryopreservation Symposium COST Action 871, Final meeting, Agrocampus Ouest INHP, Angers, pp 12–12
Reed BM, Uchendu E (2008) Controlled rate cooling. In: Reed BM (ed) Plant cryopreservation: a practical guide. Springer, New York, pp 77–92
Sakai A, Kobayashi S, Oiyama I (1990) Cryopreservation of nucellar cells of navel orange (Citrus sinensis Osb. var. brasiliensis Tanaka) by vitrification. Plant Cell Rep 9:30–33
Suszka B, Muller C, Bonnet-Masimbert M (1996) Seeds of forest broadleaves from harvest to sowing. INRA, Paris, pp 99–123
Uchendu EE, Leonard SW, Traber MG, Reed BM (2010) Vitamins C and E improve regrowth and reduce lipid peroxidation of blackberry shoot tips following cryopreservation. Plant Cell Rep 29:25–35
Uthup TK, Ravindran M, Bini K, Thakurdas S (2013) Divergent DNA methylation patterns associated with abiotic stress in Hevea brasiliensis. Mol Plant 4:996–1013
Vassar D, Mueller E, Bagga S (2009) Measurement of global DNA methylation ELISA-based method simplifies process and provides results in HTS format. Gent Eng Biotechnol News 29(7):1–3
Viejo M, Santamaría ME, Rodríguez JL, Valledor L, Meijón M, Pérez M (2012) Epi-genetics, the role of DNA methylation in tree development. In: Loyola-Vargas VM, Ochoa-Alejo N (eds) Plant cell culture methods, methods in molecular biology. Springer, Netherlands, pp 277–301
Wesley-Smith J, Walters C, Pammenter NW, Berjak P (2001a) Interactions among water content, rapid (non-equilibrium) cooling to −196°C, and survival of embryonic axes of Aesculus hippocastanum L. seeds. Cryobiology 42:196–206
Wesley-Smith J, Pammenter NW, Berjak P, Walters C (2001b) The effects of two drying rates on the desiccation tolerance of embryonic axes of recalcitrant jackfruit (Artocarpus heterophyllus Lamk.) seeds. Ann Bot 88:653–664
Wesley-Smith J, Berjak P, Pammenter NW, Walters C (2014) Intracellular ice and cell survival in cryo-exposed embryonic axes of recalcitrant seeds of Acer saccharinum: an ultrastructural study of factors affecting cell and ice structures. Ann Bot 113:695–709
Wierzbicki AT, Jerzmanowski A (2005) Suppression of histone H1 genes in Arabidopsis results in heritable developmental defects and stochastic changes in DNA methylation. Genetics 169:997–1008
Wisniewski ME, Gusta LV, Fuller MP, Karlson D (2009) Ice nucleation, propagation and deep supercooling: the lost tribes of freezing studies. In: Gusta LV, Tanino KK, Wisniewski ME (eds) Plant cold hardiness: from the laboratory to the field. CABI, Cambridge, pp 1–11
Wisniewski ME, Norelli J, Artlip T (2015) Overexpression of a peach CBF gene in apple: a model for understanding the integration of growth, dormancy, and cold hardiness in woody plants. Plant Sci 85:1–13
Zeng FS, Zhou S, Zhan YG, Dong J (2014) Drought resistance and DNA methylation of interspecific hybrids between Fraxinus mandshurica and Fraxinus americana. Trees 28:1679–1692
Zilli L, Schiavone R, Zonno V, Storelli C, Vilellaa S (2003) Evaluation of DNA damage in Dicentrarchus labrax sperm following cryopreservation. Cryobiology 47:227–235
Acknowledgments
The authors wish to thank Mariola Matelska for technical assistance. We wish to thank; eng. Anna Tyde and eng. Szymon Tyde from the Hajda-Jastrowie Forest District, for their helping of seed collections. We also thank Professor Andrzej Guranowski from Poznań University of Life Sciences, for his critical reviewing of the manuscript. This research was supported by a Grant from the National Science Centre Poland (No. N N309 101836 to PM. P), and Institute of Dendrology of the Polish Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by U. Luettge.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nuc, K., Marszałek, M. & Pukacki, P.M. Cryopreservation changes the DNA methylation of embryonic axes of Quercus robur and Fagus sylvatica seeds during in vitro culture. Trees 30, 1831–1841 (2016). https://doi.org/10.1007/s00468-016-1416-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-016-1416-3