
Abstract
Focal segmental glomerulosclerosis (FSGS) is a complex disease which describes different kinds of kidney defects, not exclusively linked with podocyte defects. Since nephrin mutation was first described in association with early-onset nephrotic syndrome (NS), many advancements have been made in understanding genetic patterns associated with FSGS. New genetic causes of FSGS have been discovered, displaying unexpected genotypes, and recognizing possible site of damage. Many recent large-scale sequencing analyses on patients affected by idiopathic chronic kidney disease (CKD), kidney failure (KF) of unknown origin, or classified as FSGS, have revealed collagen alpha IV genes, as one of the most frequent sites of pathogenic mutations. Also, recent interest in complex and systemic lysosomal storage diseases, such as Fabry disease, has highlighted GLA mutations as possible causes of FSGS. Tubulointerstitial disease, recently classified by KDIGO based on genetic subtypes, when associated with UMOD variants, may phenotypically gain FSGS features, as well as ciliopathy genes or others, otherwise leading to completely different phenotypes, but found carrying pathogenic variants with associated FSGS phenotype. Thus, glomerulosclerosis may conceal different heterogeneous conditions. When a kidney biopsy is performed, the principal objective is to provide an accurate diagnosis. The broad spectrum of phenotypic expression and genetic complexity is demonstrating that a combined path of management needs to be applied. Genetic investigation should not be reserved only to selected cases, but rather part of medical management, integrating with clinical and renal pathology records. FSGS heterogeneity should be interpreted as an interesting opportunity to discover new pathways of CKD, requiring prompt genotype–phenotype correlation. In this review, we aim to highlight how FSGS represents a peculiar kidney condition, demanding multidisciplinary management, and in which genetic analysis may solve some otherwise unrevealed idiopathic cases. Unfortunately there is not a uniform correlation between specific mutations and FSGS morphological classes, as the same variants may be identified in familial cases or sporadic FSGS/NS or manifest a variable spectrum of the same disease. These non-specific features make diagnosis challenging. The complexity of FSGS genotypes requires new directions. Old morphological classification does not provide much information about the responsible cause of disease and misdiagnoses may expose patients to immunosuppressive therapy side effects, mistaken genetic counseling, and misguided kidney transplant programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Focal segmental glomerulosclerosis (FSGS) represents one of the major causes of nephrotic syndromes (NS) and kidney failure (KF) in the USA, accounting for about 20% of NS cases in children and 40% in adults [1].
Based on data from an international survey, FSGS is predominant in North America, accounting for 19.1% of primary glomerular diseases, while it is less common in Europe, reaching about 15% among a study of 60,300 biopsy reports, resulting less common than IgA nephropathy (22.1%), as expected [2]. The increased incidence in the USA may be associated with the APOL1 risk genotype in sub-Saharan population, which is underrepresented in other countries. In a Spanish study on 9378 patients affected by NS, FSGS has been reported in 12% of cases, while membranous nephropathy (MN) is the major cause of nephrotic proteinuria in adults, with a prevalence of about 25% [3].
Consistently, in another study from China including 851 patients subjected to kidney biopsy, MN was the most frequent cause of NS (28.8%), followed by other glomerulonephritis, with FSGS affecting 5% of the patients included in the cohort [4]. FSGS is a rare disease. However, in the last 20 years, a worldwide increased prevalence of FSGS has been estimated, probably related to lifestyle and dietary habits for the secondary forms [5]. FSGS is characterized by glomerular injury, usually involving a minority of glomeruli with a segmental solidification of the tuft, deposition of extracellular matrix, and glomerular hyalinosis; moreover, light microscopy typically reveals abundant resorption of lipid droplets in the proximal tubule cells, due to heavy proteinuria. Juxtamedullary glomeruli are the more vulnerable to develop FSGS, rather than the most superficial ones, due to the higher blood flow rates and higher glomerular capillary shear-stress pressure [6]. With disease progression, the lesion may sequentially involve a higher number of glomeruli, with a more diffuse, and global, sclerosis. Overall, the definition of glomerular sclerosis and its characterization has changed considerably. In 1925, a German pathologist, Theodor Fahr, first described the histopathological features of focal segmental hyalinization in a case of “progressive lipoid nephrosis with degeneration” [7], showing association with minimal change disease (MCD), both defined as podocytopathies [8,9,10,11]. While the definition of MCD did not change much, over the years, in the mid-1980s, other patterns of glomerular damage have become part of the FSGS spectrum [12]. Even though the term “FSGS” continues to be used as an expression of a diagnosis, it is clear that it may relate to many different conditions, primary and secondary forms, and potentially hiding unexpected genotypes, as it has recently emerged [13,14,15].
FSGS usually manifests with NS, and patients can be classified as steroid-sensitive NS (SSNS) and as steroid-resistant NS (SRNS) when there is a lack of response to standard treatment with steroids and progressive kidney damage. Thus, it is frequent to consider FSGS and SRNS as synonymous [16, 17]. Monogenic forms of FSGS are more common in children with FSGS/SRNS, with a reported prevalence of about 25% [18,19,20].
Monogenic FSGS in adults is difficult to estimate because genetic testing does not represent a routine test, but is limited to selected cases with positive family history, relapse or resistance to immunosuppressive therapy, early onset of disease, or the association to extrarenal manifestations, assuming the presence of syndromic conditions. However, genetic defects leading to FSGS also happen in sporadic cases [21].
FSGS is a glomerulonephritis with a not completely clear pattern of injury and it has required special efforts for the understanding of its molecular biology and for the identification of primary and secondary causes. FSGS physiopathology has been progressively investigated, with the aim to increase scientific knowledge and to better define the critical role of the term “glomerulosclerosis” in a renal pathology scenario. The heterogeneity of FSGS and the laborious process to define the range of conditions into which the term FSGS falls have increased the complexity of defining the final and precise diagnosis, making management and treatment options more demanding. Indeed, FSGS does not represent just the description of a single disease, but rather may appear during very different mechanisms of damage. Thus, in case of kidney biopsy suggestive of a FSGS pattern, a complex explorative framework should be applied and it should definitely include genetic testing, to hopefully identify the correct cause and to apply the proper personalized treatment.
The histological definition of FSGS includes a very large disease spectrum and a morphological description is used to identify primary (idiopathic and immunological), secondary, and genetic disorders [22].
Genetic studies played a central role in the identification of genetic variants encoding proteins essential for podocyte structure and function (slit diaphragm components, actin cytoskeleton components, proteins essential for coenzyme Q10 biosynthesis, nuclear proteins, and transcription factors) that can be responsible for NS [23].
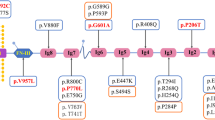
Genetic testing has progressively increased the power of discovery for hereditary forms, with more than 60 genes considered monogenic causes of FSGS/NS (Table 1). So, currently, a correct integrated approach should consider clinical data, medical history, family history, renal pathology when available, and a genomic evaluation [29]. This new path of analysis may be tough for clinicians, requiring a deep knowledge of the disease and a larger availability of diagnostic and therapeutic tools, making the genotype–phenotype correlation progressively more challenging.
Earlier genetic studies of FSGS used positional cloning mapping [25, 27] applied to large families with multiple affected family members, and targeted single gene sequencing technology to detect causal mutations in already established NS genes.
Those approaches have been useful to identify rare mutations in single genes highly expressed in podocytes and among the glomerular filtration barrier [47,48,49]. However, it traditionally required time-consuming and non-cost-effective Sanger sequencing validations, representing a sensitive technique but more problematic when applied to heterogeneous disorders caused by multiple genes [50, 51].
Thanks to the advent of recent applications of high-throughput technologies, using massive parallel sequencing, the strategies used to analyze the genomic background of patients affected by complex diseases, such as FSGS/SRNS, have completely changed [52, 53]. Next-generation sequencing (NGS) technologies include the analysis of (a) targeted panels of genes of interest (eventually selected based on hypothesis-driven approach), (b) the coding exonic regions (whole exome sequencing (WES)), or (c) the entire coding and non-coding genomic background (whole genome sequencing (WGS)). Those methods have dramatically increased the power of capturing disease-causing mutations [51], with a broader spectrum of diagnostic yield, not only including pathogenic variants in well-known NS genes, but also leading to discovery of unexpected genotypes.
Consequent to the introduction of these new tools, clinicians may investigate through larger lenses the genomic background in FSGS/SRNS patients, with promising results and new scenarios, performing a more comprehensive diagnosis. Furthermore, massive sequencing analysis has also raised the possibility of dealing with the discovery of unpredictable pathogenic mutations, leading to consideration of new genotype–phenotype correlations and the chance of thinking outside the classical pathways of disease. Unfortunately, not all centers have the availability of expensive NGS applications, thus often, genetic testing remains only applied to selected cases.
While the term “phenocopy” was introduced in medicine about 75 years ago, recently it has been subjected to conceptual expansion in human diseases [54]. A progressively increasing number of genes known to be associated with different disorders, acting as phenocopies, have emerged from sequencing data from FSGS/SRNS cohorts (Table 2).
So, if it is true that FSGS heterogeneity is well established, and if the main morphologic features are the focal fibrosis and glomerular scars, what lies behind this? How should we consider a pattern of glomerulosclerosis in this new heterogeneous fashion? For many years, the main interest in the genetics of FSGS/SRNS has been podocyte-centric, maintaining the interest of clinicians and geneticists among podocyte and glomerular filtration barrier components.
Progressively, new classes of genes have been described in cohorts of NS/FSGS patients, not directly associated with podocyte function and structure, but associated with different mechanisms of kidney damage, that may not primarily cause NS, but mimic FSGS and SRNS.
In this paper, we want to emphasize how new genetic insights have demonstrated that FSGS is a complex disease, characterized by “many masks,” and requiring a new “open vision” in its etiological investigation and clinical management. A new critical and integrated approach is needed in order to obtain the most accurate genotype–phenotype correlation.
Classification evolution: background of the FSGS issue
FSGS comprises a group of clinical-pathologic conditions clinically characterized by heavy proteinuria, hyperlipidemia, edema, and hypoalbuminemia with histological features of obliteration of glomerular capillaries by extracellular matrix with segmental distribution. However, FSGS lesions are heterogeneous. In general, primary FSGS refers to the idiopathic form with severe proteinuria, without a specific or labeled cause and for which circulating immunological triggers have been considered, even if not yet clearly identified. Secondary forms include those FSGS cases related to specific and recognizable causes, such as viral infections, drugs/toxin exposure, maladaptive nephron response, and genetic mutations.
In 2004, a working group of international renal pathologists convened at Columbia University to define and formulate the features of histological patterns of FSGS to create a histopathological classification of the disease. Columbia classification was born with the aim to define morphological criteria for the different pathological features of FSGS, for primary as well as secondary forms. It became a guide to use standardized pathological nomenclature. Five mutually exclusive morphologic variants were described, differentiating FSGS in (a) tip lesion, (b) cellular, (c) perihilar, (d) collapsing, and (e) not otherwise specified (NOS) variants (Table 3), referring to both primary and secondary forms of FSGS [59,60,61].
This morphological description led to new considerations of this disease, no longer limited to provide a pure description of kidney biopsy, but rather placed in a broader setting of new clinical-pathological scenario. Thus, the first FSGS classification was an excellent starting point to develop new future studies with the aim of understanding the molecular mechanisms differentiating FSGS variants, their different outcome and clinical progression, starting from morphological heterogeneity and trying to reduce the ambiguous use of the term “FSGS”.
While the Columbia classification aimed towards a morphological description of proliferative and sclerosing histopathological patterns with no specific correlation to pathogenesis and no contribution to treatment options, to implement the pure descriptive features with etiology and pathogenesis correlations, in 2007, the term “taxonomy” was first introduced to define an integrated and multiple-level analysis in the spectrum of heterogeneous FSGS disease [62]. The aim of the taxonomy of podocytopathies was to provide a categorization of patterns of podocyte injuries describing FSGS as a well-characterized glomerular disease due to the grade of sustained podocyte rearrangement, detachment, and apoptosis. In this new classification, the podocyte, being the main site of damage, takes the center stage, becoming the major protagonist to differentiate the kind of damage. The new approach to classification of podocytopathies distinguished four different glomerular pathways of injury, considering podocyte number modification and integrating morphological features with etiology, including idiopathic, genetic, and reactive forms. This new classification included (a) minimal change nephropathy (MCN) characterized by podocyte injury without modification of podocyte number,(b) focal segmental glomerulosclerosis (FSGS) with loss of podocytes, cell death, and insufficient repair activity; (c) diffuse mesangial sclerosis (DMS) characterized by mesangial expansion with mild proliferation, podocyte hypertrophy and hyperplasia, and lower degree of cell differentiation; and (d) collapsing glomerulopathy (CG) characterized by collapse of the glomerular tuft in at least one glomerulus with hyperplasia and hypertrophy of de-differentiated podocytes, leading to pseudo-crescent formation [62] (Table 4).
Even if the description of FSGS has gained more deep knowledge and a better diagnostic approach, over the years, its definition and classification are still subjected to evolution and updates.
The recent KDIGO guidelines, published in 2021, have proposed a new, more recent classification for FSGS that distinguishes four groups, based on light microscopy lesions, in order to improve clinical and treatment management. The aim of the new classification was to integrate a more comprehensive pathophysiology meaning and treatment options. The updated nomenclature includes (a) primary FSGS with extensive foot process effacement and sudden NS, usually linked to permeability factors still investigated and not yet well established; (b) genetic forms for all the familial, sporadic, and syndromic conditions due to pathogenic mutation in autosomal dominant or recessive or X-linked genes, known to be associated with FSGS and NS; (c) secondary forms, referring to the viral and toxic-induced FSGS cases, and including also the maladaptive conditions caused by normal or reduced nephron mass, often associated with segmental foot process effacement and milder proteinuria; and (d) FSGS of undetermined causes (FSGS-UC) in which all the other cases of unknown origin are included [63] (Table 5).
Thus, in the last 20 years, the description and the classification of FSGS have deeply evolved, from the pure morphological descriptions to an integrated classification based on podocyte fate and disease progression, until the most recent etiological classification with potential treatment options.
Nephrologists, pathologists, and scientists have focused on the role of the podocyte as the cell primarily involved in the regulation of glomerular filtration homeostasis. Therefore, a lot of advancements have been made in understanding the biology of podocyte cells and the role of genetic modifications altering the glomerular cell balance.
So far, approximately 60 genes have been described in association with podocytopathies and NS (Table 1), representing the main clinical sign of primary FSGS and also the second most common cause of chronic kidney disease (CKD) in children and young adults less than 25 years old [64].
During the first years of the twenty-first century, a common idea was that podocyte damage was involved in different forms of human and experimental glomerular disease, such as MCD, FSGS, CG, and membranous and diabetic nephropathies, all of which diseases are related to clinical manifestation of NS. Thanks to the growing interest of the research community in understanding the pathogenesis of the different forms of glomerulonephritis, today glomerular diseases have been better characterized, providing new knowledge in terms of the molecular, immunological, and genetic mechanisms.
Epithelial visceral cells directly regulate the glomerular filtration rate. Most of the diseases caused by abnormal glomerular cell function are characterized by podocyte injuries and/or dysfunction [9, 65,66,67,68]. Podocytopathies are then defined as a group of diseases, including FSGS and MCD, characterized by structural and functional podocyte impairment causing NS as an expression of glomerular filtration barrier damage. However, this classification also has been subjected to a progressive evolution, since new insights about FSGS have been achieved.
Since nephrin (NPHS1) was first described as causing early-onset NS, new observations and updated understanding have occurred. Thanks to massive parallel DNA sequencing technologies, FSGS is no longer an area of localized sclerosis associated with podocyte defects. While initially the major genes causing monogenic forms of NS/FSGS described were those associated with defects in podocytes, slit diaphragm, glomerular basement membrane (GBM), or altering actin remodeling resulting in podocyte dysfunction [37, 69, 70], in recent years, also, genes encoding proteins working far from the glomerulus have been added to the list of genes that can potentially cause FSGS. In a study from 2018, including 300 patients affected by SRNS and subjected to WES, the authors found phenocopies in 5% of the cohort. This has been the first report applying the concept of phenocopy to SRNS, explaining the possibility that FSGS may be associated with mutation in genes that do not purely affect podocytes, even if leading to proteinuria [19].
FSGS glomerular scars
In a single kidney biopsy, the features of FSGS may be wide. Kidney sampling may be tricky and sometimes it is possible that sclerotic glomeruli may be unsampled, resulting in specimens with normal glomeruli at light microscopy evaluation, but extensive foot process effacement at electron microscopy. This event should always predict the possibility of a sclerotic pattern in the glomeruli not collected. Proteinuria is one of the most common clinical findings in FSGS. Renal pathology in FSGS patients, as well as other proteinuric glomerular diseases, has demonstrated the importance of podocyte structure and glomerular barrier in the homeostasis of the urine filtration mechanism. The glomerular filtration barrier is composed of podocytes, GBM, and endothelial cells. A crosstalk between podocytes and endothelial cells exists, through the production of vascular endothelial grow factor (VEGF) by podocytes [71].
Interestingly, studies on animal models using the NEP25 chimeric mouse, in which only some podocytes express the toxin receptor human CD25, suggest that podocyte injury can extend from receptor-positive to receptor-negative podocytes, due to a different hypothesis: damaged podocytes may release toxic molecules such as chemokines, TGF-β, endothelin-1 altering podocyte survival, and also reduce the concentration of cell protective factors such as VEGF, due to altered environment after podocyte death. Reduced podocyte survival may also result from loss of podocyte–podocyte interactions or apoptosis signals through altered gap junctions, coming from damaged podocytes. Consequently, podocyte-to-podocyte damage transmission has been hypothesized as one of the mechanisms to explain the progressive extension of glomerulosclerosis, resulting from both direct and indirect triggers [72].
Glomerular scars and extracellular matrix distribution may result in the following three main events: (1) matrix deposition involves the glomerulus directly, as a response to inflammatory injuries caused by systemic inflammatory diseases with necrotizing insults, like vasculitis or lupus nephritis; (2) matrix deposition among the mesangium and GBM, usually where extracellular matrix already exists, with the aim of preserving the glomerular structures, as may happen in diabetic nephropathy and amyloidosis; (3) matrix deposition occurs in capillary loops in the setting of the glomerulus, in conditions like primary FSGS or benign nephroangiosclerosis [73].
The mechanism of glomerular scar formation is still under evaluation, but it has been described as involving cytokines such as TGF-β. TGF-β has been demonstrated to be overexpressed in glomerular disease with podocyte dysfunction [74]. The proteinuria activates molecules responsible for epithelial-to-mesenchymal trans-differentiation in the tubulo-interstitial compartment and the further generation of profibrotic cytokines and inflammatory molecules, to step through damaged podocytes [75, 76].
In addition, damaged tubular cells activate the renin angiotensin system with increased levels of angiotensin II, which acts on mesangial cells by activating them and consequently causing the production of extracellular matrix, through transcription factor “sterol-responsive element-binding protein” (SREBP-1), and finally leading to TGF-β1 upregulation with profibrogenic stimuli [77].
Schiffer et al. evaluated the role of TGF-β and SMAD family proteins in the apoptosis of podocytes and the development of glomerulosclerosis. Using TGF-β transgenic mice and cultured murine podocytes treated with TGF-β, the authors demonstrated that both TGF-β and SMAD7 cause apoptosis of the podocytes but with a different mechanism: while TGF-β activates of mitogen-activated protein (MAP) kinase p38 and classic effector caspase-3, SMAD7 inhibits the NF-κB pathway (nuclear factor kappa-light-chain-enhancer of activated B cells), enhancing the apoptotic activity of TGF-β and therefore the development and progression of glomerulosclerosis [78].
Immunohistochemistry and in situ hybridization analysis in idiopathic FSGS kidney biopsies also demonstrated the involvement of thrombospondin-1 (TSP-1), TGF-β type II receptor (TGF-βIIR) in the increased production of extracellular matrix, through SMAD signaling [79].
The role of TGF-β in the development of glomerular diseases is also evidenced by the finding of elevated TGF-β levels in urine from 42 patients with glomerulonephritis compared to 11 healthy patients, as descripted by Murakami et al. [80].
Although scars are a simple and common histological lesion in various glomerular pathologies, the mechanisms are not entirely clear. There is a complex molecular pathway, both from a biochemical and etiopathogenetic point of view, that can contribute to the development of glomerular sclerosis. With increasing use of precision medicine tools and next-generation sequencing, it has been progressively discovered how glomerular scars are not the expression of a unique exclusive event or just podocyte-related. Thus, FSGS heterogeneity can be considered an opportunity to interpret and to solve new molecular pathways able to influence the clinical manifestations of the disease. FSGS may occlude various etiopathogenetic causes that we need to explore in case of idiopathic, unknown origin, and potentially hereditary cases.
Genetics of FSGS
Table 1 lists the monogenic causes of FSGS/NS. Over 60 genes have been described as causative of FSGS/SRNS, classified based on the affected glomerular pathway.
Hereditary FSGS should be suspected when it is reported with positive family history, early-onset disease, in case of extrarenal phenotypes, and rapid decline of kidney function or lack of treatment response. When dominant genes are mutated, there is segregation of the disease through generations, while recessive forms usually show absent expression of the disease between generations, with unaffected healthy parents, being heterozygous carriers of the recessive allele or completely healthy in the case of de novo mutations. Clinicians should always investigate the presence of extra-renal manifestations, due to the possibility of syndromic genetic forms of disease, in which NS or a wide range of proteinuria could be associated, such as for example deafness, ocular abnormalities, heart defects, or other nonspecific systemic manifestations, as may happen in complex systemic disorders such as Fabry disease [81]. Incomplete penetrance and variable expression complicate this scenario, with the possibility of having asymptomatic patients while others show a wide spectrum of manifestations, from a mild phenotype with low-grade proteinuria to severe NS and its complications, to progressive CKD and KF, even when having the same genetic mutation. Very little information is available regarding the correlation between genetic FSGS and clinical/histological features. The different classes of FSGS categorize the type and grade of podocyte injuries, going from depletion to apoptosis, to de-differentiation of podocytes, but this morphological classification does not provide any information about the cause of kidney damage, the different pathways leading to disease, recognizable clinical manifestations, or prognostic orientation.
Specific correlations between genetic mutations in FSGS and renal pathology features have not yet been described. Thus, the discrimination between hereditary forms and primary FSGS is pretty difficult. The variable penetrance and expressivity in monogenic FSGS/SRNS explain the difficulty to establish when a patient with FSGS/SRNS would need genetic testing, with challenging individualization of a precise molecular diagnosis, even in the most experienced nephrology clinical setting.
The majority of NS-associated genes are autosomal recessive. NPHS1 (OMIM 602716) encodes nephrin, an immunoglobulin protein, which represents the hallmark of genetic NS/FSGS, being the first recessive gene discovered to be causative for congenital nephrotic syndrome (CNS) in the Finnish population in 1998. It is the most frequent cause of early-onset NS accounting 40 to 60% of CNS [25, 82], however, mutations in this gene may also occur in sporadic FSGS [83].
Proximal tubular dilatation may be found in kidney biopsy of patients with NPHS1 mutations [84]. Since nephrin was identified, many genes have been consequently discovered, mapping podocyte and glomerular filtration barriers.
NPHS2 (OMIM 604766) encodes for podocin, a transmembrane protein located in intracellular podocyte junctions, closely working with NPHS1 and CD2AP OMIM 604241 in the regulation of the slit diaphragm. Interestingly, a variable association has been reported between type of mutations and histological features in NPHS2-associated FSGS. While truncating variants in NPHS2 have been reported with a DMS phenotype, “less” deleterious missense mutations would be more frequently associated with an FSGS phenotype, demonstrating that renal pathology may depend upon the “developmental era” in which a specific gene mutation occurs [85]. Also, the variant p.R229Q is considered an NPHS2 polymorphism, with a high frequency (about 3%) in non-Finnish Europeans, but becoming deleterious when in compound heterozygosity with a missense NPHS2 variant in trans, if occurring between exons 7 and 8 [86].
PLCE1 OMIM 608414 encodes for phospholipase C epsilon 1, and it is one of the major causes of isolated DMS during childhood [87]. However, when PLCE1 non-truncation mutations occur, they may cause adult FSGS as a degenerative defect more than as a result of a developmental defect [27, 88]. Also, PLCE1 was recently identified as a regulator of podocyte migration and differentiation through Rho GTPase interaction [89].
LAMB2 (OMIM 150325) encodes a GBM component, working beside COLA4 heterodimers. It is one of the most common causes of isolated CNS, but also causes a syndromic form of FSGS in the context of Pierson syndrome, characterized by CNS, microcoria, and neurodevelopmental disorders [90].
While NPHS1, NPHS2, PLCE1, and LAMB2 are mostly associated with early-onset severe NS during the fetal period or first year of life, with rapid progression to KF, other recessive genes like MYO1E (OMIM 601479) are more likely associated with childhood-onset FSGS/SRNS and a later development of KF. Furthermore, MYO1E has been also associated with MCD biopsy findings [91]. As expected, recessive genetic causes are more frequently found in children, with a more severe and highly penetrant phenotype, while autosomal dominant genes are more frequently mutated in adults, with WT1 as an exception, because it is associated with a broader age of onset range of disease [92].
INF2 (OMIM 610982) is the most frequent autosomal dominant gene, responsible for 9–17% of adult familial FSGS, while TRPC6 (OMIM 603652) and ACTN4 (OMIM 604638) account for up to 12% and 3.5% of late-onset dominant familial FSGS, respectively [93,94,95,96,97]. ACTN4 is a possible cause of sporadic cases, as well [98]. INF2 encodes the inverted formin 2, involved in podocyte shape through actin cytoskeleton regulation and it is expressed in podocyte but also in heart, liver, and peripheral nerves, explaining the association with the Charcot-Marie-Tooth (CMT) neuropathy, in which FSGS is present in 75% of cases, as reported in the study from Boyer et al. [99].
TRPC6 (OMIM 603652) encodes TRP cationic channel 6, involved in calcium traffic and representing one of the major components of the slit diaphragm [100]. It works closely with the cytoskeleton resulting in regulation of podocyte migration and motility [101].
ACTN4 (OMIM 604368) encodes α-actinin-4 which provides foot processes adhesion to the GBM, leading to foot process effacement in both sporadic and familial FSGS [102, 103].
TRIM8 (OMIM 606125) is an autosomal dominant gene recently identified in a very large cohort of pediatric individuals with SRNS/FSGS and in patients with epilepsy, with most of the pathogenic truncating mutations located in the last exon of the gene, very close to the C-terminal region [104].
Syndromic FSGS may occur in case of mutations in WT1, PAX2, SMARCAL1, LMX1B, LAMB2, and COQ10-related kidney nephropathies. PAX2 (OMIM 167409) encodes a transcription factor important for brain, eye, and embryonic kidney development. PAX2 has historically been associated with Papillo-renal syndrome, characterized by congenital abnormalities of the kidney and urinary tract (CAKUT), mostly renal hypoplasia and vesicoureteral reflux (VUR), and coloboma [105]. Since 2014, it has been identified as a cause of adult-onset familial FSGS, even without congenital abnormalities or extrarenal associated phenotypes [36]. Little is known about the molecular pathway leading PAX2 to cause FSGS, but one hypothesis could be the regulation of WT1 by PAX2 [106], or a maladaptive response in case of PAX2-induced CAKUT with reduced nephron mass. Thus, PAX2 probably represents one of the first examples of phenocopy in FSGS. WT1 (OMIM 607102) is an autosomal dominant gene associated with the development of isolated Wilms tumor, isolated nephrotic proteinuria, or in the setting of syndromic conditions like Denys–Drash syndrome (DDS) and Frasier syndromes (FS), both including FSGS in association with sexual abnormalities [107].
APOL1 OMIM 603743 is a common gene following recessive Mendelian trait, frequently mutated in a specific subpopulation. APOL1 is an interesting gene with a high frequency of mutation in African Americans with sub-Saharan ancestry, leading to a three- to fourfold increased risk of developing FSGS and a twofold increased risk of developing KF. While APOL1 variants confer protection from sleeping sickness, high-risk genotypes (G1-G1, G1-G2, G2-G2) increase the risk of developing glomerulosclerosis, as demonstrated by transgenic mice with podocyte-specific expression of APOL1 G1/G2 alleles which develop proteinuria, foot process effacement, and FSGS. APOL1 high-risk genotype is also associated with viral infections such as HIV, COVID-19, and malaria [108,109,110,111].
Hidden phenocopies behind FSGS
COL4A spectrum disorders
A large proportion of unknown CKD and KF may hide a genetic disease–causing defect [112].
A progressively increasing number of studies have disclosed how FSGS/SRNS may start from genetic mutations in genes far different from those classically defined as “podocyte-related.” FSGS may be difficult to differentiate from Alport syndrome (AS), based just upon pathology findings and symptoms, and it has been demonstrated by analysis of genetic insights that AS may often be mislabeled as FSGS [113, 114].
AS is an inherited glomerular disorder caused by pathogenic mutations of collagen alpha 4 genes (COL4A3 (MIM: 203780; 104200; 620320), COL4A4 (MIM 203780; 141200), COL4A5 (MIM 301050). It is the most common glomerular inherited disorder. Hematuria, hearing loss, and progressive KF are the most typical symptoms related to AS. It has been estimated that in Europe, untreated patients affected by X-linked AS may rapidly evolve to progressive KF with a median age of 22 years [115], with males strongly affected and showing a more severe phenotype than females, in whom a less severe and variable phenotype is more common, due to X-chromosome inactivation (lyonizations) [116, 117].
Collagen IV represents the most abundant protein found in the GBM, and it strongly brings together podocytes and endothelial cells in the proper function of the glomerular filtration barrier. About 80% of AS patients may carry an X-linked mechanism of inheritance involving the COL4A5 gene, with high penetrance of hematuria in males, showing the most severe phenotype. About 15% of patients with AS may show a recessive mode of inheritance due to mutations in COL4A3 or COL4A4, both located on chromosome 2. However, a small portion of individuals, often underdiagnosed, and accounting for about 5% of AS patients, may show milder clinical manifestations with an autosomal dominant pattern of disease where just one mutated copy of COL4A3, COL4A4 is identified; these patients are frequently defined as patients affected by thin basement membrane disease (TBMD) [116, 118,119,120]. Also, digenic inheritance has been reported [29, 121].
More than five thousand pathogenic variants have been recognized in COL4A3, COL4A4, and COL4A5, with 50% of missense variants affecting glycine residues, 20% of which variants are truncating nonsense mutations and frameshifts, while 15% respectively are large indels and deletions or variants altering the splicing mechanism [122].
Interestingly, COL4A3, COL4A4, and COL4A5 pathogenic variants have been found in patients with persistent proteinuria or SRNS associated with FSGS, in both children and adult populations [123, 124]. Indeed, when large cohorts of CKD patients have been subjected to genotyping through WES or WGS, a high incidence of collagen IV pathogenic variants was found [125]. Interestingly, many recent reports and studies reveal that collagen IV genes are becoming the most common monogenic cause of FSGS in adults [126, 127].
So it is more common now to talk about COLA4-spectrum disorders, more than AS-related disease. Barua et al. performed WES in 193 patients with familial and sporadic forms of FSGS, using a gene panel of 109 genes related to FSGS, NS, CAKUT, and nephronophthisis. Pathogenic mutations in 28% of patients with a positive family history and 11% for sporadic cases were reported. Overall, the diagnostic yield for definitely pathogenic variants reached 11% of the total cohort, while 9% were likely pathogenic mutations. Interestingly, more than half (55%) of the pathogenic variants involved all the three collagen IV genes, COL4A3, COL4A4, and COL4A5, usually implicated in AS [128]. In another study from the Columbia University group, where one of the largest cohorts was sequenced, including 3315 patients affected by CKD, Groopmanan et al. identified monogenic disorders in 10% of the cohort, of those, about 100 patients, accounting for 30% of the diagnostic yield, showed mutations in COL4A3, COL4A4, or COL4A5. This study demonstrated that collagen IV variants were the second most frequent genetic disorders in the CKD cohort, after the 31% of PKD1 and PKD2 pathogenic variants. Only 35 out of 91 patients (38%) with diagnostic variants of collagen IV had a clinical diagnosis of AS or TBMD [129].
These findings demonstrate that collagen IV gene mutations represent one of the leading genetic causes of masked FSGS, often unrecognized, suggesting the importance of genetic screening in clinical practice. Genotype–phenotype correlation should be considered as a powerful tool to properly deliver tailored diagnoses, relative personalized treatment, and follow-up.
Lysosome storage dysfunction
Lysosomal storage disorders can cause podocyte damage, mimicking histological features of FSGS. Alterations in genes encoding for lysosome proteins are responsible for Fabry disease, cystinosis, Nieman-Pick disease, and Tay-Sachs disease all characterized by kidney involvement [130].
Podocytes do not have the ability to proliferate; thus, intracellular homeostasis is important for their integrity. Lysosomes are essential organelles for the survival of podocytes, for their digestive and recycling properties [15].
Among lysosomal storage diseases, Fabry disease (FD) is an X-linked disorder, caused by mutation of the GLA (MIM 301500) gene, with defect of the enzymatic activity of the α-galactosidase enzyme (α-GalA), leading to abnormal and excessive deposition of neutral glycosphingolipids, including globotriaosylceramide (Gb3) in endothelial, epithelial, and smooth muscle cells. Progressive accumulation of glycosphingolipids causes clinical abnormalities of kidney, heart, skin, eye, brain, and peripheral nervous system. The accumulation of glycosphingolipids in renal lysosomes causes a progressive worsening of kidney function often resulting in KF [131].
In the early stages of FD, patients may show difficulties in concentrating urine, together with non-nephrotic proteinuria and modest hypertension, finally leading to impaired kidney function often resulting in KF in the third to fifth decades of life [132]. FD is therefore a multisystem and progressive disease.
FD may show histological features of FSGS. The morphologic alterations are determined by Gb3 deposits in all components of kidney parenchyma: glomerular, tubular, interstitial, and vascular. The deposits are observed in visceral podocytes earlier than in the Bowman’s capsule epithelium, in mesangial cells, in endothelial cells of glomeruli and peritubular capillaries, in the smooth muscle cells of arteries, in tubular cells, and most frequently in the distal tract. Interstitial cells are rarely involved. In advanced cases of disease, there are signs of segmental or global glomerulosclerosis, interstitial fibrosis, tubular atrophy, and arteriosclerosis [133]. The lysosomal deposits are lamellar electron dense structures (intercalated with electron-lucid lamellas), commonly termed “zebra bodies,” or “myelin figures” visible at the electron microscopy analysis of kidney biopsies. However, although electron microscopy is very useful to recognize Gb3 deposits associated with FD, they can be observed also in other conditions, such as silica nephropathy and pseudolipidosis, caused by the use of drugs such as amiodarone, chloroquine, and hydroxychloroquine.
Trimarchi et al. described the significant impact of electron microscopy in the specific differential diagnosis of FD in a patient initially classified as having FSGS by the analysis of kidney tissue only by light microscopy. They found lamellar electron dense lipids, as zebra bodies, under examination with electron microscopy in this 37-year-old patient initially treated with steroids as having FSGS for a long time. The correct diagnosis of FD allowed them to start the correct enzymatic replacement therapy [134]. The development of glomerular sclerosis in FD would seem to be mediated by an inflammatory state due to the deposition of Gb3 in the tissues. The increase of cytokines such as TGF-ß would therefore be responsible [135].
Data deriving from studies on the immune system of patients with FD are very interesting. Lymphocytes, monocytes, and granulocytes of patients with FD express more adhesion molecules than those in the healthy population [136]. Furthermore, Gb3 activates Toll-like receptor 4 (TLR4) that stimulates immune cells through Notch1 and the NF-κB transcription factors, with release of proinflammatory and profibrotic cytokines [137]. TGF-ß is crucial for fibrotic damage in response to chronic inflammation in FD, determining the synthesis of extracellular matrix in kidney cells via epithelial-to-mesenchymal transition. Indeed, deposition of Gb3 in glomerular cells is followed by FSGS until global glomerular sclerosis [138]. Studies of urinary proteomics revealed the presence of fibroblast growth factor 23, uromodulin, and podocalyxin in patients with FD, responsible for an inflammatory state and the activation of the fibrosis pathway in these patients. Enzyme replacement therapy can reduce the inflammatory state by reducing Gb3 deposits, only if administered in the early stages of FD. A late onset of enzyme replacement therapy is less effective on renal pathology, when fibrogenesis processes have already begun.
Another syndrome characterized by lysosomal anomalies is action myoclonus–renal failure syndrome (AMRF). It is an autosomal recessive progressive myoclonus epilepsy (PME) associated with kidney dysfunction, caused by loss-of-function mutations in the SCARB2 (MIM 254900) gene encoding lysosomal integral membrane protein type 2 (LIMP2). This very rare syndrome appears in the second or third decade of life. LIMP2 traffics β-glucocerebrosidase to the lysosomal membrane. Mutations lead to glucosylceramide accumulation and neurologic symptoms including progressive action myoclonus, seizures, and ataxia [139]. Kidney involvement in AMRF consists of proteinuria that can evolve to NS, and even development of KF [140].
Badhwar et al. in 2004 described 15 cases with AMRF, all patients showing proteinuria, detected between age 9 and 30. The kidney biopsies performed in these patients showed collapsing FSGS. SCARB2/LIMP2 mutation also causes failure of endosomes containing reabsorbed proteins to fuse with lysosomes in the proximal tubular epithelial cells, with development of tubular proteinuria [141].
There are other lysosomal dysfunction diseases characterized by kidney impairment, mainly due to alteration of the proximal tubular compartment, with Fanconi syndrome, low molecular weight proteinuria, and even progressive KF. Cystinosis, Dent disease, and Lowe syndrome are due to genetic defects responsible for severe kidney damage. KF can be explained by the development of tubulointerstitial fibrosis [142].
Renal lipid dysregulation is furthermore one of the factors responsible for the development of diabetic nephropathy.
Tubulointerstitial disease
Since the KDIGO consensus conference in 2015, different subclasses of autosomal dominant tubulointerstitial kidney disease (ADTKD) have been classified based on the genetic background [143].
Among these genes, UMOD (OMIM 191845) is a gene encoding uromodulin (also known as Tamm-Horsfall protein) that is the most abundant protein in normal urine. Uromodulin is essential in the regulation of ion transport, immunomodulation, protection against urinary tract infections, and prevention of the formation of kidney stones and oxidative stress [144, 145].
UMOD gene mutations are known to be related to ADTKD, also known as ADTKD-UMOD, which may slowly progress to CKD, leading to KF [146].
Gast et al. [147] analyzed patients with CKD stages 3–5, in order to identify patients with inherited kidney disease. They observed that ADTKD-UMOD was the most common genetic form of kidney disease after autosomal dominant polycystic kidney disease.
Moreover, Groopman et al. [129], conducting exome sequencing and diagnostic analysis in patients affected by CKD, identified 66 distinct monogenic disorders, and found that 3% were explained by mutations in UMOD, in a very large cohort of 3315 CKD patients [129]. Under a clinical profile, about 80% of patients affected by ADTKD-UMOD presented hyperuricemia that starts before the progressive loss of kidney function and is the main symptom of the disease. Additionally, gout and medullary renal cysts are sometimes present. ADTKD-UMOD is a difficult condition to diagnose, requiring a high clinical suspicion and confirmation by genetic testing. The urinary sediment is bland with absent to mild albuminuria or proteinuria and no hematuria. Patients with UMOD mutation usually develop KF between the third and sixth decade of life, whereas the onset of gout occurs between the ages of 3 and 51 years [147].
Renal pathology is usually unspecific, and patients affected by ADTKD-UMOD may be mislabeled as FSGS [22]. Electron microscopy may describe fibrillary intracellular deposits of uromodulin, stored within endoplasmic reticulum in tubular cells of Henle’s loop, explaining the frequently defective urine-concentrating process [148].
Thus, in patients with histological diagnosis of FSGS in whom an underlying secondary cause of FSGS is suspected, it is necessary to obtain a correct medical and family history for gout or kidney disease (FSGS of unclear etiology) and testing serum urate levels and urine analysis. In case of a strong clinical suspicion of ADTKD-UMOD, genetic tests are recommended to detect any mutations in UMOD gene.
CLCN5 (OMIM 300008) is an X-linked recessive gene expressed in proximal tubules and collecting duct. It is responsible for a rare syndromic condition called Dent disease type 1 (Dent-1), characterized by hypercalciuria, nephrocalcinosis, kidney stone development, CKD, and progression to KF in which tubular proteinuria occurs. Sometimes proteinuria may reach nephrotic range values and it may be mistaken for a glomerular defect [149], and a glomerulosclerosis phenotype is possible [150]. The hypothesis is that CLCN5 may cause FSGS and NS through regulation of podocyte trafficking, in addition to tubular dysfunction [151], so the effective molecular targets of CLCN5 have not yet been fully clarified. Also, mutations in OCRL (OMIM 300535) may cause a severe tubular dysfunction called Lowe syndrome in the setting of Dent disease type 2 (Dent-2), characterized by ocular abnormalities, intellectual impairment, CKD, and rapid progression to KF, in which persistent proteinuria and FSGS have been described, as well [152]. Thus, CLCN5 and OCRL should be taken into consideration as potential phenocopies of FSGS, in a genetic setting.
Ciliopathy
Ciliopathy identifies a group of genetic disorders characterized by retinal degeneration, cerebral abnormalities, and kidney dysfunction and frequently presenting nephronophthisis (NPHP), a recessive condition frequently leading to CKD in young adults [43].
Many genes have been identified as disease-causing in NPHP [153]. However, three genes have been implicated in FSGS reports.
TTC21B (OMIM 612014) encodes for IFT139, an intra-flagellar transport-A component located at the primary cilium of young podocytes, while in adults in non-ciliated podocytes IFT139 is subjected to redistribution along the intracellular microtubule compartment. While TTC21B had been initially recognized as a potential genetic cause of NPHP (OMIM 613820), and short-rib thoracic dysplasia 4 with or without polydactyly it has also been reported as a possible genetic cause of glomerular compartment defects, in addition to tubulointerstitial alterations, manifesting FSGS [154,155,156].
CC2D2A (OMIM 612013) encodes a ciliary protein which works as a barrier to restrict protein flow between the ciliary membrane and plasma. Recently, a compound heterozygous missense mutation in CC2D2A has been reported in a girl affected by NPHP and FSGS [157].
NPHP4 (OMIM 606966) is a recessive gene causing Senior-Loken syndrome 4 [158] and it has been identified in a single consanguineous family with segregation of proteinuria and kidney phenotype in multiple siblings, with a single patient undergoing kidney biopsy and diagnosed with FSGS [159]. The mechanism of disease causing FSGS through NPHP genes remains unexplained, but it is possibly a secondary adaptive response to nephron loss or podocyte cytoskeleton dysfunction in TTC21B mutations.
Conclusions
About 10% of the population affected by CKD has a monogenic disorder [160, 161].
CKD is a complex disease, with different molecular mechanisms responsible for progressive kidney function decline. Patients affected by progressive CKD may show nonspecific histopathological features at kidney biopsy, such as a wide spectrum of glomerulosclerosis, interstitial fibrosis, and tubular atrophy that can be due to different pathogenic mechanisms. Thus, in a simplistic view, glomerulosclerosis may represent both a sign of progression of chronic inflammation and kidney injury, as well as a renal pathology hallmark in the diagnosis of FSGS, remaining an unspecific sign, detectable in different renal diseases.
It has been estimated that about 25% of dialyzed patients are classified as patients affected by KF of unknown origin. Thanks to the integration of DNA sequencing and genotyping approaches in kidney diseases, it has been demonstrated that a large proportion of patients with KF may remain unclassified, eventually hiding a genetic disease–causing defect [112].
Among these patients, FSGS, whose incidence is growing [3,4,5], represents a very heterogenous and complex disease. The recent updated KDIGO classification suggested the importance of identifying the underlying cause of primary, secondary, and genetic FSGS, required for personalized clinical management and treatment options.
So far, over 60 genes have been identified as monogenic causes of FSGS. FSGS and SRNS are frequently used synonymously due to the lack of immunosuppressive response especially in adults. Podocyte genes are commonly mutated in both familial and sporadic cases, but recent insights obtained from massive sequencing analysis on large cohorts of CKD patients have demonstrated that new patterns of injury need to be investigated as phenocopies in FSGS.
FSGS/SRNS management needs a new updated framework, which should consider an integrated approach between phenotype characterization, pathophysiology, and genetic testing to properly identify the correct causes of disease and to specifically drive treatment options, avoiding side effects and complications. Genetic versus non-genetic etiologies of SRNS and FSGS may have different prognosis, especially during childhood and in those resistant cases eventually planning a living donor transplant. Genetic testing is needed for familiar screening to determine donor eligibility status and to identify unsuitable potential familiar donors carrying one of the known genetic variants [22]. Thus, NGS should become a diagnostic standard.
Collagen IV genes including COLA4A3, COL4A4, and COL4A5, usually associated with hereditary forms of Alport syndrome, represent the emerging most frequent cause of FSGS in patients with otherwise unknown CKD or KF. Moreover, the growing interest in rare complex diseases, such as Fabry disease, has revealed that FSGS may hide mutations in the GLA gene leading to lysosomal dysfunction, manifesting glomerulosclerosis features at the kidney level. Even if glomerular and tubulointerstitial compartments seem to be separate sites of damage, some of the genes regulating tubular homeostasis and cilia structure may show a sort of dualism. UMOD, CLCN5, OCRL, NPHP4, and TTC21B may cause tubulointerstitial diseases such as ADTKD, NPHP, or Dent disease, but they are now included in the genetic panels for genetic screening of patients affected by FSGS, as they can phenocopy it. Many other new genes classically involved in syndromic/non-syndromic disorders, have been identified in sequencing analysis of patients showing FSGS phenotype.
In conclusion, new insights into FSGS heterogeneity represent an opportunity, because it moves the attention from podocytes to other areas of interest, discovering new potential triggers of damage, manifesting with proteinuria and glomerular scars. The incomplete penetrance and pleiotropic expression of FSGS/SRNS require a broader genetic analysis in order to provide a tailored and targeted diagnosis and for treatment selection.
Glomerular scars are not a specific and distinctive sign of FSGS; however, they represent the hallmark in the diagnosis of this proteinuric disease. In addition, FSGS classification has been subjected to rearrangements, and new monogenic causes of FSGS are discovered on a monthly basis. When we look at a kidney biopsy specimen through the lens of a light microscope, we cannot understand what is hidden behind glomerular scars, but we can just describe the captured features. An integrated approach that includes patient “phenotyping,” renal pathology, clinical reports, and sequencing analysis is now mandatory to interpret the data and to offer the better diagnosis and management to patients affected by kidney diseases, in the era of precision medicine.
References
Kitiyakara C, Kopp JB, Eggers P (2003) Trends in the epidemiology of focal segmental glomerulosclerosis. Semin Nephrol 23:172–182
O’Shaughnessy MM, Hogan SL, Thompson BD et al (2018) Glomerular disease frequencies by race, sex and region: results from the International Kidney Biopsy Survey. Nephrol Dial Transplant 33:661–669
Rivera F, Lopez-Gomez JM, Perez-Garcia R et al (2004) Clinicopathologic correlations of renal pathology in Spain. Kidney Int 66:898–904
Jin B, Zeng C, Ge Y et al (2014) The spectrum of biopsy-proven kidney diseases in elderly Chinese patients. Nephrol Dial Transplant 29:2251–2259
Schwimmer JA, Markowitz GS, Valeri AM et al (2003) Secondary focal segmental glomerulosclerosis in non-obese patients with increased muscle mass. Clin Nephrol 60:233–241
Remuzzi A, Mazerska M, Gephardt GN et al (1995) Three-dimensional analysis of glomerular morphology in patients with subtotal nephrectomy. Kidney Int 48:155–162
Henke F Handbuch der speziellen pathologischen Anatomie und Histologie. J. Springer, Berlin 1924. VOL. II.
D’Agati VD (2012) Pathobiology of focal segmental glomerulosclerosis: new developments. Curr Opin Nephrol Hypertens 21:243–250
Pollak MR (2002) Inherited podocytopathies: FSGS and nephrotic syndrome from a genetic viewpoint. J Am Soc Nephrol 13:3016–3023
IJpelaar DHT, Farris AB, Goemaere N et al (2008) Fidelity and evolution of recurrent FSGS in renal allografts. J Am Soc Nephrol 19:2219–2224
Di Leo V, Capaccio F, Gesualdo L (2020) Preeclampsia and Glomerulonephritis: A Bidirectional Association. Curr Hypertens Rep 22:36
Rich AR (1957) A hitherto undescribed vulnerability of the juxtamedullary glomeruli in lipoid nephrosis. Bull Johns Hopkins Hosp 100:173–186
Bose B, Cattran D, Toronto Glomerulonephritis R (2014) Glomerular diseases: FSGS. Clin J Am Soc Nephrol 9:626–632
Sethi S, Glassock RJ, Fervenza FC (2015) Focal segmental glomerulosclerosis: towards a better understanding for the practicing nephrologist. Nephrol Dial Transplant 30:375–384
Li G, Kidd J, Li PL (2020) Podocyte lysosome dysfunction in chronic glomerular diseases. Int J Mol Sci 21:1559
Lee JM, Kronbichler A, Shin JI et al (2021) Current understandings in treating children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 36:747–761
Reidy K, Kaskel FJ (2007) Pathophysiology of focal segmental glomerulosclerosis. Pediatr Nephrol 22:350–354
Nagano C, Yamamura T, Horinouchi T et al (2020) Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Sci Rep 10:270
Warejko JK, Tan W, Daga A et al (2018) Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 13:53–62
Park E, Lee C, Kim NKD et al (2020) Genetic study in korean pediatric patients with steroid-resistant nephrotic syndrome or focal segmental glomerulosclerosis. J Clin Med 9:2013
Santin S, Bullich G, Tazon-Vega B et al (2011) Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 6:1139–1148
Chun J, Wang M, Wilkins MS et al (2020) Autosomal dominant tubulointerstitial kidney disease-uromodulin misclassified as focal segmental glomerulosclerosis or hereditary glomerular disease. Kidney Int Rep 5:519–529
Preston R, Stuart HM, Lennon R (2019) Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how? Pediatr Nephrol 34:195–210
Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, Unanue ER, Shaw AS. (2003) CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 300(5623):1298-300. https://doi.org/10.1126/science.1081068
Kestila M, Lenkkeri U, Mannikko M et al (1998) Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell 1:575–582
Boute N, Gribouval O, Roselli S et al (2000) NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24:349–354
Hinkes B, Wiggins RC, Gbadegesin R et al (2006) Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38(12):1397–1405
Kaplan JM, Kim SH, North KN et al (2000) Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24:251–256
Lipska-Zietkiewicz BS (1993) In: Adam MP et al (eds) Genetic steroid-resistant nephrotic syndrome overview, in GeneReviews((R)), Seattle (WA)
Gee HY, Saisawat P, Ashraf S et al (2013) ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 123:3243–3253
Brown EJ, Schlondorff JS, Becker DJ et al (2010) Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 42:72–76
Gee HY, Zhang F, Ashraf S et al (2015) KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest 125:2375–2384
Mele C, Iatropoulos P, Donadelli R et al (2011) MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 365:295–306
Winn MP, Conlon PJ, Lynn KL et al (2005) A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308:1801–1804
Gee HY, Ashraf S, Wan X et al (2014) Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet 94:884–890
Barua M, Stellacci E, Stella L et al (2014) Mutations in PAX2 associate with adult-onset FSGS. J Am Soc Nephrol 25:1942–1953
Ha TS (2017) Genetics of hereditary nephrotic syndrome: a clinical review. Korean J Pediatr 60:55–63
Has C, Sparta G, Kiritsi D et al (2012) Integrin alpha3 mutations with kidney, lung, and skin disease. N Engl J Med 366:1508–1514
Ashraf S, Gee HY, Woerner S et al (2013) ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest 123:5179–5189
Diomedi-Camassei F, Di Giandomenico S, Santorelli FM et al (2007) COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 18:2773–2780
Heeringa SF, Chernin G, Chaki M et al (2011) COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest 121:2013–2024
Lopez LC, Schuelke M, Quinzii CM et al (2006) Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 79:1125–1129
Braun DA, Schueler M, Halbritter J et al (2016) Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int 89:468–475
Boerkoel CF, Takashima H, John J et al (2002) Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 30:215–220
Becherucci F, Landini S, Cirillo L et al (2020) Sound alike: phenocopies in steroid-resistant nephrotic syndrome. Int J Environ Res Public Health 17:8363
Hermle T, Schneider R, Schapiro D et al (2018) GAPVD1 and ANKFY1 Mutations Implicate RAB5 Regulation in Nephrotic Syndrome. J Am Soc Nephrol 29:2123–2138
Benoit G, Machuca E, Antignac C (2010) Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol 25:1621–1632
Joshi S, Andersen R, Jespersen B et al (2013) Genetics of steroid-resistant nephrotic syndrome: a review of mutation spectrum and suggested approach for genetic testing. Acta Paediatr 102:844–856
Sampson MG, Hodgin JB, Kretzler M (2015) Defining nephrotic syndrome from an integrative genomics perspective. Pediatr Nephrol 30:51–63 (quiz 59)
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467
Neveling K, Feenstra I, Gilissen C et al (2013) A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 34:1721–1726
Domingo-Gallego A, Pybus M, Bullich G et al (2022) Clinical utility of genetic testing in early-onset kidney disease: seven genes are the main players. Nephrol Dial Transplant 37:687–696
Vivante A, Hildebrandt F (2016) Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12:133–146
Grebe H (1954) Etiological role of the genes and phenocopies in human abnormalities. Acta Genet Med Gemellol (Roma) 3:197–209
Mochizuki T, Lemmink HH, Mariyama M et al (1994) Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 8:77–81
Castelletti F, Donadelli R, Banterla F et al (2008) Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci U S A 105:2538–2543
Kantarci S, Al-Gazali L, Hill RS et al (2007) Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet 39:957–959
Attree O, Olivos IM, Okabe I et al (1992) The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 358:239–242
D’Agati VD, Fogo AB, Bruijn JA et al (2004) Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 43:368–382
Stokes MB, D’Agati VD (2014) Morphologic variants of focal segmental glomerulosclerosis and their significance. Adv Chronic Kidney Dis 21:400–407
D’Agati VD, Kaskel FJ, Falk RJ (2011) Focal segmental glomerulosclerosis. N Engl J Med 365:2398–2411
Barisoni L, Schnaper HW, Kopp JB (2007) A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol 2:529–542
Disease K (2021) Improving global outcomes glomerular diseases work, G., KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int 100:S1–S276
Sadowski CE, Lovric S, Ashraf S et al (2015) A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26:1279–1289
Adler S (1992) Characterization of glomerular epithelial cell matrix receptors. Am J Pathol 141:571–578
Simons M, Schwarz K, Kriz W et al (2001) Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am J Pathol 159:1069–1077
Schwarz K, Simons M, Reiser J et al (2001) Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest 108:1621–1629
Reiser J, Kriz W, Kretzler M et al (2000) The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol 11:1–8
Zenker M, Machuca E, Antignac C (2009) Genetics of nephrotic syndrome: new insights into molecules acting at the glomerular filtration barrier. J Mol Med (Berl) 87:849–857
Lovric S, Ashraf S, Tan W et al (2016) Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 31:1802–1813
Eremina V, Cui S, Gerber H et al (2006) Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol 17:724–735
Matsusaka T, Sandgren E, Shintani A et al (2011) Podocyte injury damages other podocytes. J Am Soc Nephrol 22:1275–1285
Faraggiana T, Giannakakis C (2008) Glomerulosclerosis: pathogenetic mechanisms and possibility of regression. G Ital Nefrol 25(Suppl 44):S27–S32
Lee HS (2012) Mechanisms and consequences of TGF-ss overexpression by podocytes in progressive podocyte disease. Cell Tissue Res 347:129–140
Liu Y (2004) Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 15:1–12
Liu Y (2010) New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol 21:212–222
Wang TN, Chen X, Li R et al (2015) SREBP-1 mediates angiotensin II-induced TGF-beta1 upregulation and glomerular fibrosis. J Am Soc Nephrol 26:1839–1854
Schiffer M, Bitzer M, Roberts IS et al (2001) Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest 108:807–816
Kim JH, Kim BK, Moon KC et al (2003) Activation of the TGF-beta/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int 64:1715–1721
Murakami K, Takemura T, Hino S et al (1997) Urinary transforming growth factor-beta in patients with glomerular diseases. Pediatr Nephrol 11:334–336
de Haan A, Morel CF, Eijgelsheim M et al (2023) Fabry disease with atypical phenotype identified by massively parallel sequencing in early-onset kidney failure. Clin Kidney J 16:722–726
Machuca E, Benoit G, Nevo F et al (2010) Genotype-phenotype correlations in non-finnish congenital nephrotic syndrome. J Am Soc Nephrol 21(7):1209–1217
Zhuo L, Huang L, Yang Z et al (2019) A comprehensive analysis of NPHS1 gene mutations in patients with sporadic focal segmental glomerulosclerosis. BMC Med Genet 20:111
Saleem MA (2019) Molecular stratification of idiopathic nephrotic syndrome. Nat Rev Nephrol 15:750–765
DeMars PA, Fleming JD, Benham PA (1991) Ethics across the occupational therapy curriculum. Am J Occup Ther 45:782–787
Tory K, Menyhard DK, Woerner S et al (2014) Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet 46:299–304
Gbadegesin R, Hinkes BG, Hoskins BE et al (2008) Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol Dial Transplant 23:1291–1297
Hildebrandt F, Heeringa SF (2009) Specific podocin mutations determine age of onset of nephrotic syndrome all the way into adult life. Kidney Int 75:669–671
Yu S, Choi WI, Choi YJ et al (2020) PLCE1 regulates the migration, proliferation, and differentiation of podocytes. Exp Mol Med 52:594–603
Matejas V, Hinkes B, Alkandari F et al (2010) Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat 31:992–1002
Al-Hamed MH, Al-Sabban E, Al-Mojalli H et al (2013) A molecular genetic analysis of childhood nephrotic syndrome in a cohort of Saudi Arabian families. J Hum Genet 58:480–489
Chernin G, Vega-Warner V, Schoeb DS et al (2010) Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clin J Am Soc Nephrol 5:1655–1662
Morales-Alvarez MC, Knob A, Rennke HG et al (2022) Clinical and pathological heterogeneity in FSGS due to INF2 mutations. Kidney Int Rep 7:2741–2745
Barua M, Brown EJ, Charoonratana VT et al (2013) Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int 83:316–322
Boyer O, Benoit G, Gribouval O et al (2011) Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol 22:239–245
Weins A, Kenlan P, Herbert S et al (2005) Mutational and biological analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol 16:3694–3701
Caridi G, Lugani F, Dagnino M et al (2014) Novel INF2 mutations in an Italian cohort of patients with focal segmental glomerulosclerosis, renal failure and Charcot-Marie-Tooth neuropathy. Nephrol Dial Transplant 29(Suppl 4):iv80-6
Bartram MP, Habbig S, Pahmeyer C et al (2016) Three-layered proteomic characterization of a novel ACTN4 mutation unravels its pathogenic potential in FSGS. Hum Mol Genet 25:1152–1164
Boyer O, Nevo F, Plaisier E et al (2011) INF2 mutations in charcot-marie-tooth disease with glomerulopathy. N Engl J Med 365:2377–2388
Reiser J, Polu KR, Moller CC et al (2005) TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37:739–744
Farmer LK, Rollason R, Whitcomb DJ et al (2019) TRPC6 binds to and activates calpain, independent of its channel activity, and regulates podocyte cytoskeleton, cell adhesion, and motility. J Am Soc Nephrol 30:1910–1924
Smoyer WE, Mundel P, Gupta A et al (1997) Podocyte alpha-actinin induction precedes foot process effacement in experimental nephrotic syndrome. Am J Physiol 273(1 Pt 2):F150–F157
Henderson JM, Alexander MP, Pollak MR (2009) Patients with ACTN4 mutations demonstrate distinctive features of glomerular injury. J Am Soc Nephrol 20:961–968
Weng PL, Majmundar AJ, Khan K et al (2021) De novo TRIM8 variants impair its protein localization to nuclear bodies and cause developmental delay, epilepsy, and focal segmental glomerulosclerosis. Am J Hum Genet 108:357–367
Bower M, Salomon R, Allanson J et al (2012) Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum Mutat 33:457–466
Yang Y, Jeanpierre C, Dressler GR et al (1999) WT1 and PAX-2 podocyte expression in Denys-Drash syndrome and isolated diffuse mesangial sclerosis. Am J Pathol 154:181–192
Lipska-Zietkiewicz BS (1993) In: Adam MP et al (eds) WT1 Disorder, in GeneReviews((R)), Seattle (WA)
Genovese G, Friedman DJ, Ross MD et al (2010) Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329:841–845
Friedman DJ, Pollak MR (2011) Genetics of kidney failure and the evolving story of APOL1. J Clin Invest 121:3367–3374
Shetty AA, Tawhari I, Safar-Boueri L et al (2021) COVID-19-associated glomerular disease. J Am Soc Nephrol 32:33–40
Beckerman P, Bi-Karchin J, Park AS et al (2017) Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 23:429–438
Quaglia M, Musetti C, Ghiggeri GM et al (2014) Unexpectedly high prevalence of rare genetic disorders in kidney transplant recipients with an unknown causal nephropathy. Clin Transplant 28:995–1003
Comic J, Riedhammer KM, Gunthner R et al (2022) The multifaceted phenotypic and genotypic spectrum of type-IV-collagen-related nephropathy-A human genetics department experience. Front Med (Lausanne) 9:957733
Lieberman KV, Chang AR, Block GA et al (2022) The KIDNEYCODE Program: diagnostic yield and clinical features of individuals with CKD. Kidney360 3:900–909
Temme J, Kramer A, Jager KJ et al (2012) Outcomes of male patients with Alport syndrome undergoing renal replacement therapy. Clin J Am Soc Nephrol 7:1969–1976
Jais JP, Knebelmann B, Giatras I et al (2000) X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol 11:649–657
Bekheirnia MR, Reed B, Gregory MC et al (2010) Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol 21:876–883
Longo I, Porcedda P, Mari F et al (2002) COL4A3/COL4A4 mutations: from familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int 61:1947–1956
Kashtan CE (2005) Familial hematurias: what we know and what we don’t. Pediatr Nephrol 20:1027–1035
Antignac C (1995) Molecular genetics of basement membranes: the paradigm of Alport syndrome. Kidney Int Suppl 49:S29–S33
Savige J, Renieri A, Ars E et al (2022) Digenic alport syndrome. Clin J Am Soc Nephrol 17:1697–1706
Savige J, Huang M, Croos Dabrera MS et al (2022) Genotype-phenotype correlations for pathogenic COL4A3-COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant alport syndrome. Front Med (Lausanne) 9:865034
Voskarides K, Damianou L, Neocleous V et al (2007) COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol 18:3004–3016
Gast C, Pengelly RJ, Lyon M et al (2016) Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 31:961–970
Lata S, Marasa M, Li Y et al (2018) Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med 168:100–109
Malone AF, Phelan PJ, Hall G et al (2014) Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int 86:1253–1259
Braunisch MC, Buttner-Herold M, Gunthner R et al (2018) Heterozygous COL4A3 variants in histologically diagnosed focal segmental glomerulosclerosis. Front Pediatr 6:171
Yao T, Udwan K, John R et al (2019) Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clin J Am Soc Nephrol 14:213–223
Groopman EE, Marasa M, Cameron-Christie S et al (2019) Diagnostic utility of exome sequencing for kidney disease. N Engl J Med 380:142–151
Platt FM, d’Azzo A, Davidson BL et al (2018) Lysosomal storage diseases. Nat Rev Dis Primers 4:27
Branton MH, Schiffmann R, Sabnis SG et al (2002) Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 81:122–138
Schiffmann R, Hughes DA, Linthorst GE et al (2017) Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 91:284–293
Abensur H, Reis MA (2016) Renal involvement in Fabry disease. J Bras Nefrol 38:245–254
Trimarchi H, Karl A, Rana MS et al (2013) Initially nondiagnosed fabry’s disease when electron microscopy is lacking: the continuing story of focal and segmental glomerulosclerosis. Case Rep Nephrol Urol 3:51–57
Lee MH, Choi EN, Jeon YJ et al (2012) Possible role of transforming growth factor-beta1 and vascular endothelial growth factor in Fabry disease nephropathy. Int J Mol Med 30:1275–1280
Rozenfeld P, Agriello E, De Francesco N et al (2009) Leukocyte perturbation associated with Fabry disease. J Inherit Metab Dis 32(Suppl 1):S67–S77
Feriozzi S, Rozenfeld P (2021) Pathology and pathogenic pathways in fabry nephropathy. Clin Exp Nephrol 25:925–934
Rozenfeld P, Feriozzi S (2017) Contribution of inflammatory pathways to fabry disease pathogenesis. Mol Genet Metab 122:19–27
Dibbens L, Schwake M, Saftig P et al (2016) SCARB2/LIMP2 deficiency in action myoclonus-renal failure syndrome. Epileptic Disord 18:63–72
Caridi G, Trivelli A, Sanna-Cherchi S et al (2010) Familial forms of nephrotic syndrome. Pediatr Nephrol 25:241–252
Desmond MJ, Lee D, Fraser SA et al (2011) Tubular proteinuria in mice and humans lacking the intrinsic lysosomal protein SCARB2/Limp-2. Am J Physiol Renal Physiol 300:F1437–F1447
Surendran K, Vitiello SP, Pearce DA (2014) Lysosome dysfunction in the pathogenesis of kidney diseases. Pediatr Nephrol 29:2253–2261
Laura Econimo CS, Zeni L, Cortinovis R, Alberici F, Rampoldi L, Scolari F, Izzi C (2022) Autosomal dominant tubulointerstitial kidney disease: an emerging cause of genetic CKD. Kidney Int Rep 7:2332–2344
Devuyst O, Olinger E, Rampoldi L (2017) Uromodulin: from physiology to rare and complex kidney disorders. Nat Rev Nephrol 13:525–544
LaFavers KA, Macedo E, Garimella PS et al (2019) Circulating uromodulin inhibits systemic oxidative stress by inactivating the TRPM2 channel. Sci Transl Med 11:eaaw3639
Bollee G, Dahan K, Flamant M et al (2011) Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol 6:2429–2438
Gast C, Marinaki A, Arenas-Hernandez M et al (2018) Autosomal dominant tubulointerstitial kidney disease-UMOD is the most frequent non polycystic genetic kidney disease. BMC Nephrol 19:301
Scolari F, Caridi G, Rampoldi L et al (2004) Uromodulin storage diseases: clinical aspects and mechanisms. Am J Kidney Dis 44:987–999
Beara-Lasic L, Cogal A, Mara K et al (2020) Prevalence of low molecular weight proteinuria and Dent disease 1 CLCN5 mutations in proteinuric cohorts. Pediatr Nephrol 35:633–640
Wang X, Anglani F, Beara-Lasic L et al (2016) Glomerular pathology in dent disease and its association with kidney function. Clin J Am Soc Nephrol 11:2168–2176
Solanki AK, Arif E, Morinelli T et al (2018) A novel CLCN5 mutation associated with focal segmental glomerulosclerosis and podocyte injury. Kidney Int Rep 3:1443–1453
Kaneko K, Hasui M, Hata A et al (2010) Focal segmental glomerulosclerosis in a boy with Dent-2 disease. Pediatr Nephrol 25:781–782
Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. N Engl J Med 364:1533–1543
Bullich G, Vargas I, Trujillano D et al (2017) Contribution of the TTC21B gene to glomerular and cystic kidney diseases. Nephrol Dial Transplant 32:151–156
Olinger E, Phakdeekitcharoen P, Caliskan Y et al (2022) Biallelic variants in TTC21B as a rare cause of early-onset arterial hypertension and tubuloglomerular kidney disease. Am J Med Genet C Semin Med Genet 190:109–120
Huynh Cong E, Bizet AA, Boyer O et al (2014) A homozygous missense mutation in the ciliary gene TTC21B causes familial FSGS. J Am Soc Nephrol 25:2435–2443
Awazu M, Yamada M, Asada N et al (2022) A girl with a mutation of the ciliary gene CC2D2A presenting with FSGS and nephronophthisis. CEN Case Rep 11:116–119
Otto E, Hoefele J, Ruf R et al (2002) A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet 71:1161–1167
Mistry K, Ireland JH, Ng RC et al (2007) Novel mutations in NPHP4 in a consanguineous family with histological findings of focal segmental glomerulosclerosis. Am J Kidney Dis 50:855–864
Devuyst O, Knoers NV, Remuzzi G et al (2014) Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet 383:1844–1859
Mallett A, Patel C, Salisbury A et al (2014) The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis 9:98
Funding
Open access funding provided by Università degli Studi di Bari Aldo Moro within the CRUI-CARE Agreement. All the authors are members of the European Reference Network for Rare Kidney Diseases (ERKNet).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mitrotti, A., Giliberti, M., Di Leo, V. et al. Hidden genetics behind glomerular scars: an opportunity to understand the heterogeneity of focal segmental glomerulosclerosis?. Pediatr Nephrol 39, 1685–1707 (2024). https://doi.org/10.1007/s00467-023-06046-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-023-06046-1