
Abstract
Background
Primary hyperoxalurias (PHs) constitute rare disorders resulting in abnormal glyoxalate metabolism. PH-associated phenotypes range from progressive nephrocalcinosis and/or recurrent urolithiasis to early kidney failure.
Methods
A retrospective study was conducted for patients with confirmed PH diagnoses from three tertiary centers in Saudi Arabia. Detailed clinical molecular diagnosis was performed for 25 affected individuals. Whole exome sequencing (WES)–based molecular diagnosis was performed for all affected individuals.
Results
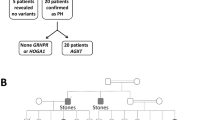
The male:female ratio was 52% male (n = 13) and 48% female (n = 12), and consanguinity was present in 88%. Nephrolithiasis and/or nephrocalcinosis were present in all patients. Kidney stones were present in 72%, nephrocalcinosis in 60%, hematuria in 32%, proteinuria in 16%, abdominal pain in 36%, developmental delay in 8%, and chronic kidney disease stage 5 (CKD stage 5) was observed in 28% of the patients. The most common PH disorder was type I caused by variants in the AGXT gene, accounting for 56%. The GRHPR gene variants were identified in 4 patients, 16% of the total cases. Seven patients did not reveal any associated variants. Missense variants were the most commonly observed variants (48%), followed by frame-shift duplication variants (28%).
Conclusions
Characterization of the genetic and clinical aspects of PH in this unique population provides direction for improved patient management and further research.
Graphical abstract

A higher resolution version of the Graphical abstract is available as Supplementary information
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary hyperoxalurias (PHs) are rare inborn errors of metabolism (glyoxylate) which lead to the overproduction of oxalate in the liver [1, 2]. The accumulation of oxalate results in the calcium oxalate (CaOx) crystals that are deposited in the urinary tract and kidneys [3]. Hyperoxalurias are classified into two main categories (PHs and SHs) [4, 5]. Secondary hyperoxalurias (SHs) generate from an increased oxalate absorption due to various alterations, including intestinal inflammatory diseases, very low calcium diet, bariatric surgery, or undue oxalate precursors intake [6, 7].
PH type I (OMIM 604,285) is the most severe type of hyperoxaluria. It is an autosomal recessive disease resulting in an insufficiency of alanine glyoxylate aminotransferase enzyme (AGT), which takes part in glyoxylate metabolism final step [8]. As a result, glyoxylate is not effectively metabolized to glycine, causing overproduction of both glycolate and oxalate [9]. PH type I is associated with progressive nephrocalcinosis, or recurrent urolithiasis, which leads to CKD stage 5 during the first 2–3 decades of life [10]. Infantile oxalosis is the most severe clinical presentation of PH type I.
PH type II (OMIM 260,000) results from the deficiency of the enzyme glyoxylate reductase/hydroxypyruvate reductase (GRHPR), which plays a key role in the gluconeogenic pathway and helps in the removal of cytosolic glyoxylate [6, 11]. Lack of GRHPR results in glyoxylate and hydroxypyruvate accumulation that is processed by lactate dehydrogenase to L-glycerate and oxalate. The clinical picture of PH type II is milder compared to PH type I; however, affected individuals may suffer from frequent episodes of urolithiasis that might lead to kidney failure [12].
PH III (OMIM 613,616) results from recessive variants in the HOGA1 gene, which encodes the enzyme 4-hydroxy-2-oxoglutarate aldolase (HOGA). Clinical presentations include a less severe form with early onset, a low chronic kidney disease risk, an early disease onset, impaired kidney function and recurrent stone disease [13].
In this study, we sought to characterize the genetic and clinical characteristics of PH patients in Saudi Arabia in order to provide guidance to clinicians and researchers interested in this rare disorder.
Materials and methods
Inclusion criteria
The study included cases with confirmed molecular diagnoses having PHs type I, II, or III. In addition, cases that presented pathogenic or likely pathogenic variants in either AGXT, GRHPR, or HOGA1 genes according to the ACMG criteria were included. Finally, affected individuals having a variant of uncertain significance (VUS) were also included, who had PHs before molecular testing and who had consistent PH phenotypes.
Genetic testing
The molecular diagnosis in PHs patients was performed using single gene testing or WES, as previously reported [14,15,16].
Variant classifications and prioritization
ACMG guidelines were followed for variant classification [17]. Variant filtration was performed using standard methods [18]. Different online databases (ExAC, gnomAD, 1000 genomes, etc.) were used to exclude the polymorphic nature of the identified variants. In addition, the variants were fully segregated within available family members.
Molecular biogenetic testing
Sanger sequencing was performed to segregate the identified variant in all members of the families, followed by WES. Primers were designed using Primer3 and Sanger sequencing was performed using standard methods.
Pathogenicity of the variants
Sanger sequencing confirmed the segregation of all the discovered variants. For missense variants, in silico analysis tools, including Polyphen2, SIFT, Varsome and MutationTaster were used to predict the pathogenicity of the novel variants. Conservation of the variants was investigated using HomoloGene (https://www.ncbi.nlm.nih.gov/homologene).
Results
This retrospective chart review study includes 25 patients diagnosed with PHs from three tertiary centers in Riyadh and Jeddah, Saudi Arabia (October 2019–October 2021). The IRB of KAIMRC, KSU-Medical City Hospital, (Riyadh), and KAU (Jeddah) KSA approved the study. Since the study is based on retrospective data, written informed consent was not required.
Demographic details of patients
The study consisted of 25 patients with 13 males and 12 females, and affected individuals ranged from 3 to 50 years of age (Table 1). The majority (96%) of participants (24/25) were less than 18 years old. All of the affected individuals were from different regions of Saudi Arabia [North (7/25: 28%), central (13/25: 52%), West (5/25: 20%)]. Consanguinity was reported in 88% of the cases (22/25). The age at diagnosis of the affected individuals was between 6 months and 39 years, and a positive family history was observed in 32% (8/25) of cases.
Initial symptoms
In most cases, the affected individuals presented with vomiting, flank pain, abdominal pain, kidney stones, hematuria, fever, and nephrocalcinosis.
Developmental milestones
Of the 25 patients evaluated, 2 (8%) manifested failure to thrive and developmental delay, including delayed gross and fine motor development, and language issues. All of the other affected individuals achieved normal developmental milestones. Three of the affected individuals were deceased (3/25; 12%) at the ages of 13 years, 39 months and 11 months. Cause of death included burden of CKD stage 5 for the youngest patient — the patient’s parents chose to give the child palliative care at home. Thus, the patient died at home with no documented hospital death report. The second patient died due to septic shock, and the third died at the age of 11 months having primary hyperoxaluria and Down syndrome.
Kidney examination
Detailed clinical investigation showed kidney manifestations in all of the affected individuals, including nephrocalcinosis, hematuria, and multiple kidney stones (Table 1). Nephrolithiasis and/or nephrocalcinosis were present in all patients. Kidney stones were present in (18/25; 72%), while nephrocalcinosis was present in (15/25; 60%), hematuria (8/25; 32%), proteinuria (4/25; 16%), and abdominal pain (9/25; 36%). The age of the patients when kidney stones were observed ranged between 22 months and 13 years, and most of the patients suffered from bilateral kidney stones (18/25; 72%). CKD stage 5 was observed in 28% of the patients (7/25). Skeletal manifestations affected 24% (6/25) of the patients, while other manifestations included recurrent urinary tract infection (12%; 3/25) (Tables 1 and 2).
Laboratory examination
Detailed laboratory investigations were performed as summarized in Table 1. Urinary oxalate concentration was investigated in 6 affected individuals (upper normal limit 0.5 mmol/L): all had elevated urinary oxalate level. Plasma oxalate concentration (normal range: 1.3–3.1 μmol/L) was measured in 10 affected individuals and all had high plasma oxalate levels. Three of the affected individuals showed both proteinuria and hematuria, and three affected individuals showed hypercalciuria (Table 1).
Other abnormalities
Two patients revealed additional clinical phenotypes such as hepatomegaly, skeletal malformation and hypotonia (2/25; 8%).
Molecular analysis
All of the 25 affected individuals were subjected to single gene testing or whole-exome sequencing (WES) using standard methods, followed by segregation analysis using Sanger sequencing [15]. Molecular genetic testing revealed a confirmed molecular diagnosis in 19/25 cases (76%), and the majority of them were previously reported disease-causing variants. Nearly all identified variants were homozygous and thus demonstrated an autosomal recessive pattern. In addition, one proband revealed an AGXT-compound heterozygous variant.
AGXT gene variants [c.33dup; p.(Lys12GlnfsX156), c.187G > C; p.(Gly63Arg)] were observed in 14 cases (56%), GRHPR gene variants [c.707A > T; p.(Glu236Val)] were observed in 4 patients (16%), an already reported HOGA1 gene variant (c.860G > T; p.Gly287Val) was observed in 1 patient (4%), and six patients had negative molecular genetics results (24%). The homozygous variant identified in the GRHPR gene (c.707A > T; p.(Glu236Val) was novel. The most frequently identified variants were missense (12/25; 48%), followed by frame-shift duplication variant (7/25; 28%) (Fig. 1). Due to clinical heterogeneity, no clear genotype–phenotype correlations could be concluded. All the identified variants in patients were also screened in all the available family members using standard Sanger sequencing.

Variants identified in the present study
The patients with kidney stones and negative molecular results were diagnosed based on the occurrence of kidney stones, type of kidney stones (Ca-oxalate in stone analysis), elevated urine glycolic and oxalic acids in urine organic acid, and elevated urine oxalate /creatinine ratio (hyperoxalemia).
Management
Dialysis was performed for seven patients (7/25; 28%). Six patients were on hemodialysis (HD), and one was initially on peritoneal dialysis then shifted to HD once referred to tertiary hospital. Depending on feasibility, HD was performed almost 5–6 sessions per week for all patients. Every patient was prescribed different treatments depending on the disease severity and condition, including pyridoxine, polystra tacrolimus, amlodipine, atenolol, cholecalciferol, colchicine, folic acid, hydrochlorothiazide, acetaminophen, prednisolone, and potassium citrate (Table 2). These medications improved the overall quality of life of the affected individuals. In addition, liver transplant was performed for two affected individuals (2/25; 8%) and the eGFR values at the time of liver transplant for patient 1 and patient 3 were 7 ml/min/1.73 m2 and 91 ml/min/1.73 m2, respectively.
Vitamin B6 (pyridoxine or pyridoxal phosphate) can help to reduce the production of oxalate in some patients affected with PH1 by enhancing the activity of the pyridoxal phosphate-dependent enzyme alanine: glyoxylate aminotransferase (AGT) [19]. Around 30–50% of PH1-affected individuals are pyridoxine-responsive, and after a minimum of 3 months, therapy resulted in a 30% reduction in urine oxalate concentration [20]. For deficient AGT to respond to pyridoxine, some degree of enzyme activity must be preserved that occurs with certain pathogenic AGXT variants [21]. Most pyridoxine-responsive individuals show maximum benefit at a dose of 5–8 mg/kg/day [22, 23]. Some require higher doses depending on the disease severity; however, a starting pyridoxine dose of 5 mg/kg/day is recommended. The conditions required for pyridoxine follow-up urine collection include stepwise increase in pyridoxine dose (maximum 10–20 mg/kg/day) [24, 25]. Vitamin B6 response can be difficult to determine in patients with CKD at advanced stage since, at that time, the urinary oxalate excretion rates may be influenced by the low GFR.
Discussion
Herein, we present the results of a retrospective study of a pediatric cohort of patients with PH from Saudi Arabia. Genetic variants were identified in 76% of patients (19/25). All patients with PH become symptomatic within the first decade of life. The majority of the PH type I affected individuals developed symptoms by 5 years of age; 1 to 2% of infants with PH type I developed kidney failure [26]. On the other hand, 3.2 years to 8.0 years was the average age for the onset of PH type II [12, 27].
While in European studies, PH type II is estimated to range from 1 to 3 in 1,000,000 people [19], the prevalence of PHs in Saudi Arabia is unknown. PH type I is estimated to have a 1:149,000 prevalence in Europeans and 1:157,000 in African Americans [28]. There is no estimated prevalence for PH type III; however, its prevalence is lower than PH type I and type II, making genotype–phenotype correlations difficult.
PHs type I–III are recessively inherited glyoxylate metabolism disorders, associated with an increased endogenetic oxalate synthesis and thus cause excessive accumulation of glyoxylate in the body. Most cases (70–80%) are classified as PH type I [24], caused by disease-causing variants in the AGXT gene that results in the absence or deficiency of the AGT enzyme. As transamination of glyoxylate to glycine is catalyzed by AGT, its deficiency allows reduction of glyoxylate into glycolate and this is then oxidized into oxalate. PH type II is less severe, resulting from the deficiency of GRHPR enzyme [1, 12, 24]. Garrelfs et al. [29] reported 101 patients from eleven countries exhibiting PH type II. A total of 82.8% of the patients in their cohort revealed urolithiasis and molecular diagnosis revealed 18 novel GRHPR gene mutations. Eight patients had CKD stage 2 and 22 showed CKD stage 5. Fifteen kidney transplantations were also performed in 11 patients [12]. PH type III is caused by disease-causing variants in HOGA1, encoding HOGA responsible for mobilizing the last step in hydroxyproline metabolism [19, 24,25,26,27,28,29,30].
The disease‐causing variants identified in our study are located in three different genes that have been previously implicated in causing PHs. According to the molecular study results, PH type I (OMIM 259,900) caused by homozygous variants in the AGXT gene (OMIM 604,285) was the most common gene mutated. The most common variant and probably the hotspot variant in this region is the frameshift variant in AGXT [c.33dup; p.(Lys12Glnfs*156)].
The present study reports 19 variants in 25 different individuals. The rate of previously reported variants associated with PH was high, while the GRHPR novel variant (c.707A > T; p.(Glu236Val) causing PH type II was identified in 4 different affected individuals. All of the identified variants in patients were also screened in all the available family members using standard Sanger sequencing. The pathogenic effect of the identified variants was confirmed using in silico prediction tools such as PolyPhen, MutationTaster, Varsome, etc. Additionally, the frequency of these variants in the general population was checked in ExAC, gnomAD, 1000 Genome Project databases, and + 2000 exomes/genomes. Additionally, segregation analysis of all the identified variants was performed using Sanger sequencing.
The phenotype in the patients presented here was mild to severe compared to previously reported cases from Saudi Arabia [31]. Indeed, the 16 patients previously reported presented with nephrolithiasis (13/16) and nephrocalcinosis (12/16). Among them, 4 patients revealed CKD stage 5, 3 underwent kidney transplantation, 4 showed nephrocalcinosis and nephrolithiasis and 3 with hyperoxaluria were asymptomatic [31]. In addition, they also observed urinary tract infection, abdominal pain, hematuria, and none of the patients presented an infantile form of PHI [31]. Furthermore, disease-causing AGXT variants [c.33dup; (p.Lys12GlnfsX156), c.187G > C; p.(Gly63Arg)] previously reported, and variants in patients presented here do not cluster within one domain and are distributed in different domains, which might be one of the reasons for heterogeneity in clinical presentation. Although, these regions are crucial for the normal function of AGXT and might be intolerant to different missense and frameshift variations reported in these regions. Consanguineous marriages are very common in Saudi Arabia, accounting for more than 52% of all marriages. Similarly, in the present study consanguineous marriages were observed in 88% of the patients. Interestingly, both consanguineous and non-consanguineous families revealed homozygous variants, showing that the variants are prevalent and hotspots in the Saudi population. The age of the affected individuals at first symptoms was between 1 month and 20 years, and the age at diagnosis was between 6 months and 39 years. In the cohort reported here, the average delay in diagnosis was approximately 3 years and 2 months.
Affected individuals diagnosed with any PHs should be treated immediately with high fluid intake. Urinary CaOx supersaturation can be reduced with alkali citrate treatment [32]. In the liver, citrate is converted to bicarbonate, and the alkali reduces the citrate reabsorption (intratubular) and thus citrate excretion is increased. For PH type I patients, the second treatment strategy includes enhancing the reduced activity of AGT [27]. As a small amount of urinary oxalate is derived from the diet in these patients, dietary oxalate restriction showed limited benefit [33]. In addition, individuals with PH1 have an excellent safety profile with pyridoxine (vitamin B6) treatment.
It was recently reported that the RNA interference (RNAi) therapeutic agent lumasiran targets the glycolate oxidase and thus reduces hepatic oxalate production. It has been reported to reduce urinary oxalate excretion, which is the main cause of kidney failure in PH1 patients. Most patients who received lumasiran treatment revealed normal levels within 6 months [34].
Such studies also highlight the need for periodic reevaluation of variants, either by molecular reanalysis, a certified laboratory, a geneticist, or scientists responsible for variant interpretation. The ACMG has issued standard guidelines for variant classification. A reasonable approach might be to review the molecular findings every 6 to 10 months if there is any reclassification of inherited VUS or likely pathogenic variant. This might help parents planning to have more children through pre-implantation genetic diagnosis (PGD) [35]. Additionally, population-specific allele identification will assist in data sharing among labs and ultimately decrease the burden. Molecular testing results are dynamic and significantly change with new variant reclassification over time that might help reduce VUS burden; however, a significant number of patients with inconclusive test results remain.
Physicians in KSA face many issues associated with disease management and treatment of rare genetic disorders. As the frequency of rare genetic disorders in KSA is more than in any other country, there is a need for a nationwide campaign that might raise awareness. KSA requires its own evidence-based nationwide guidelines for treating such rare disorders and physicians should be equipped with the latest knowledge about the latest therapies and new treatments approved by the FDA. However, prenatal genetic screening including noninvasive prenatal testing (NIPT), preimplantation genetic testing for aneuploidy (PGT-A), and preimplantation genetic testing for monogenic disorders (PGT-M) has been established recently in KSA to tackle such severe rare diseases [35, 36]. However, nationwide systematic neonatal screening and proper awareness is the need of the day [14].
Although we believe this study is the largest cohort from the region, it is limited by the relatively small sample size and the absence of functional studies. Nevertheless, we present clinical, molecular, and genetic data for 25 patients along with identification of common disease-causing variants found in the Saudi population. Such studies might help better understand the disease pathogenesis, which may facilitate evidence-based and well-designed therapies in the future.
Data availability
Upon reasonable request to the corresponding author.
References
Hoppe B (2012) An update on primary hyperoxaluria. Nat Rev Nephrol 8:467–475. https://doi.org/10.1038/nrneph.2012.113
Martin-Higueras C, Torres A, Salido E (2017) Molecular therapy of primary hyperoxaluria. J Inherit Metab Dis 40:481–489. https://doi.org/10.1007/s10545-017-0045-3
Dindo M, Conter C, Oppici E, Ceccarelli V, Marinucci L, Cellini B (2019) Molecular basis of primary hyperoxaluria: clues to innovative treatments. Urolithiasis 47:67–78. https://doi.org/10.1007/s00240-018-1089-z
Salido E, Pey AL, Rodriguez R, Lorenzo V (2012) Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta 1822:1453–1464. https://doi.org/10.1016/j.bbadis.2012.03.004
Williams EL, Acquaviva C, Amoroso A, Chevalier F, Coulter-Mackie M, Monico CG, Giachino D, Owen T, Robbiano A, Salido E, Waterham H, Rumsby G (2009) Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Hum Mutat 30:910–917. https://doi.org/10.1002/humu.21021
Cregeen DP, Williams EL, Hulton S, Rumsby G (2003) Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat 22:497. https://doi.org/10.1002/humu.9200
Bouzidi H, Majdoub A, Daudon M, Najjar MF (2016) Primary hyperoxaluria: a review. Nephrol Ther 12:431–436. https://doi.org/10.1016/j.nephro.2016.03.005
Alfadhel M, Alhasan KA, Alotaibi M, Khalid Al Fakeeh KA (2012) Extreme intrafamilial variability of Saudi brothers with primary hyperoxaluria type 1. Ther Clin Risk Manag 8:373–376. https://doi.org/10.2147/TCRM.S34954
Danpure CJ (2004) Molecular aetiology of primary hyperoxaluria type 1. Nephron Exp Nephrol 98:e39–e44
Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO (2006) Primary hyperoxaluria type 1: still challenging! Pediatr Nephrol 21:1075–1081. https://doi.org/10.1007/s00467-006-0124-4
Hoppe B, Dittlich K, Fehrenbach H, Plum G, Beck BB (2011) Reduction of plasma oxalate levels by oral application of Oxalobacter formigenes in 2 patients with infantile oxalosis. Am J Kidney Dis 58:453–455. https://doi.org/10.1053/j.ajkd.2011.05.012
Garrelfs SF, Rumsby G, Peters-Sengers H (2019) Patients with primary hyperoxaluria type 2 have significant morbidity and require careful follow-up. Kidney Int 96:1389–1399. https://doi.org/10.1016/j.kint.2019.08.018
Martin-Higueras C, Garrelfs SF, Groothoff JW, Jacob DE, Moochhala SH, Bacchetta J, Acquaviva C, Zaniew M, Sikora P, Beck BB, Hoppe B (2021) A report from the European Hyperoxaluria Consortium (OxalEurope) Registry on a large cohort of patients with primary hyperoxaluria type 3. Kidney Int 100:621–635. https://doi.org/10.1016/j.kint.2021.03.031
Alfadhel M, Umair M, Almuzzaini B, Alsaif S, AlMohaimeed SA, Almashary MA, Alharbi W, Alayyar L, Alasiri A, Ballow M, AlAbdulrahman A, Alaujan M, Nashabat M, Al-Odaib A, Altwaijri W, Al-Rumayyan A, Alrifai MT, Alfares A, AlBalwi M, Tabarki B (2019) Targeted SLC19A3 gene sequencing of 3000 Saudi newborn: a pilot study toward newborn screening. Ann Clin Transl Neurol 6:2097–2103. https://doi.org/10.1002/acn3.50898
Alfadhel M, Almuqbil M, Al Mutairi F, Umair M, Almannai M, Alghamdi M, Althiyab H, Albarakati R, Bashiri FA, Alshuaibi W, Ba-Armah D, Saleh MA, Al-Asmari A, Faqeih E, Altuwaijri W, Al-Rumayyan A, Balwi MA, Ababneh F, Alswaid AF, Eyaid WM, Almontashiri NAM, Alhashem A, Hundallah K, Bertoli-Avella A, Bauer P, Beetz C, Alrifai MT, Alfares A, Tabarki B (2021) The leukodystrophy spectrum in Saudi Arabia: epidemiological, clinical, radiological, and genetic data. Front Pediatr 9:633385. https://doi.org/10.3389/fped.2021.633385
Alfadhel M, Abadel B, Almaghathawi H, Umair M, Rahbeeni Z, Faqeih EA, Almannai M, Alasmari A, Saleh M, Eyaid W, Alfares A, Mutairi FA (2022) HMG-CoA lyase deficiency: a retrospective study of 62 Saudi patients. Front Genet 13:880464. https://doi.org/10.3389/fgene.2022.880464
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Umair M, Khan MF, Aldrees M, Nashabat M, Alhamoudi KM, Bilal M, Alyafee Y, Al Tuwaijri A, Aldarwish M, Al-Rumayyan A, Alkhalaf H, Wadaan MAM, Alfadhel M (2021) Mutated VWA8 is associated with developmental delay, microcephaly, scoliosis and play a novel role in early development and skeletal morphogenesis in Zebrafish. Front Cell Dev Biol 9:736960. https://doi.org/10.3389/fcell.2021.736960
Cochat P, Groothoff J (2013) Primary hyperoxaluria type 1: practical and ethical issues. Pediatr Nephrol 28:2273
Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, Fargue S, Groothoff J, Harambat J, Hoppe B, Jamieson NV, Kemper MJ, Mandrile G, Marangella M, Picca S, Rumsby G, Salido E, Straub M, van Woerden CS (2012) Primary hyerpoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 27:1729–1736
Fargue S, Knight J, Holmes RP, Rumsby G, Danpure CJ (2016) Effects of alanine:glyoxylate aminotransferase variants and pyridoxine sensitivity on oxalate metabolism in a cell-based cytotoxicity assay. Biochim Biophys Acta 1862:1055–1062
Monico CG, Olson JB, Milliner DS (2005) Implications of genotype and enzyme phenotype in pyridoxine response of patients with type I primary hyperoxaluria. Am J Nephrol 25:183–188
Hoyer-Kuhn H, Kohbrok S, Volland R, Franklin J, Beck BB, Hoppe B (2014) Vitamin B6 in primary hyperoxaluria 1: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 9:468–477
Bobrowski AE, Langman CB (2008) The primary hyperoxalurias. Semin Nephrol 28:152–162
Hoppe B, Beck B, Milliner DS (2009) The primary hyperoxalurias. Kidney Int 75:1264–1271
Harambat J, van Stralen KJ, Espinosa L, Groothoff JQ, Hulton SA, Cerkausiene R, Schaefer F, Verrina E, Jager KJ, Cochat P (2012) Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol 7:458–465
Leumann E, Hoppe B (2001) The primary hyperoxalurias. J Am Soc Nephrol 12:1986–1993
Hopp K, Cogal AG, Bergstralh EJ, Seide BM, Olson JB, Meek AM, Lieske JC, Milliner DS, Harris PC, Rare Kidney Stone Consortium (2015) Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol 26:2559–2570
Monico CG, Persson M, Ford GC, Rumsby G, Milliner DS (2002) Potential mechanisms of marked hyperoxaluria not due to primary hyperoxaluria I or II. Kidney Int 62:392–400
Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, Monico CG, Feinstein S, Ben-Shalom E, Magen D, Weissman I, Charon C, Frishberg Y (2010) Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet 87:392–399
Sanjad SA, Al-Abbad A, Al-Sabban E (1999) Primary hyperoxaluria type 1: an underestimated cause of nephrocalcinosis and chronic renal failure in Saudi Arabian children. Ann Saudi Med 19:4–7. https://doi.org/10.5144/0256-4947.1999.4
Leumann E, Hoppe B, Neuhaus T (1993) Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 7:207–211
Sikora P, von Unruh GE, Beck B, Feldkötter M, Zajaczkowska M, Hesse A, Hoppe B (2008) [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int 73:1181–1186
Garrelfs SF, Frishberg Y, Hulton SA, Koren MJ, O’Riordan WD, Cochat P (2021) Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med 384:1216–1226. https://doi.org/10.1056/NEJMoa2021712
Alyafee Y, Alam Q, Tuwaijri AA, Umair M, Haddad S, Alharbi M, Alrabiah H, Al-Ghuraibi M, Al-Showaier S, Alfadhel M (2021) Next-generation sequencing-based Pre-Implantation Genetic Testing for Aneuploidy (PGT-A): first report from Saudi Arabia. Genes (Basel) 12:461. https://doi.org/10.3390/genes12040461
Alyafee Y, Al Tuwaijri A, Alam Q, Umair M, Haddad S, Alharbi M, Ballow M, Al Drees M, AlAbdulrahman A, Al Khaldi A, Alfadhel M (2021) Next generation sequencing based Non-invasive Prenatal Testing (NIPT): first report from Saudi Arabia. Front Genet 12:630787. https://doi.org/10.3389/fgene.2021.630787
Acknowledgements
We thank the families for their cooperation.
Funding
This study was funded by KAIMRC.
Author information
Authors and Affiliations
Contributions
Alfadhel M. Umair M. Alhasan KA: Writing the manuscript, submitting the manuscript, patient management and clinical diagnosis. Alghamdi MA, Fakeeh KA, Al Qahtani AT, Shalaby, MA, Farahat A, Kari JA: Reviewing the data, data collection, clinical diagnosis and patient management. Raina R, Cochat P: reviewing the data, and approving the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alfadhel, M., Umair, M., Alghamdi, M.A. et al. Clinical and molecular characterization of a large primary hyperoxaluria cohort from Saudi Arabia: a retrospective study. Pediatr Nephrol 38, 1801–1810 (2023). https://doi.org/10.1007/s00467-022-05784-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-022-05784-y