
Abstract
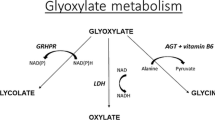
Primary hyperoxaluria type 1, the most common form of primary hyperoxaluria, is an autosomal recessive disorder caused by a deficiency of the liver-specific enzyme alanine: glyoxylate aminotransferase (AGT). This results in increased synthesis and subsequent urinary excretion of the metabolic end product oxalate and the deposition of insoluble calcium oxalate in the kidney and urinary tract. As glomerular filtration rate (GFR) decreases due to progressive renal involvement, oxalate accumulates and results in systemic oxalosis. Diagnosis is still often delayed. It may be established on the basis of clinical and sonographic findings, urinary oxalate ± glycolate assessment, DNA analysis and, sometimes, direct AGT activity measurement in liver biopsy tissue. The initiation of conservative measures, based on hydration, citrate and/or phosphate, and pyridoxine, in responsive cases at an early stage to minimize oxalate crystal formation will help to maintain renal function in compliant subjects. Patients with established urolithiasis may benefit from extracorporeal shock-wave lithotripsy and/or JJ stent insertion. Correction of the enzyme defect by liver transplantation should be planned, before systemic oxalosis develops, to optimize outcomes and may be either sequential (biochemical benefit) or simultaneous (immunological benefit) liver–kidney transplantation, depending on facilities and access to cadaveric or living donors. Aggressive dialysis therapies are required to avoid progressive oxalate deposition in established end-stage renal disease (ESRD), and minimization of the time on dialysis will improve both the patient’s quality of life and survival.
Similar content being viewed by others

References
Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N, on behalf of the Société de Néphrologie and the Société de Néphrologie Pédiatrique (1995) Epidemiology of primary hyperoxaluria type 1. Nephrol Dial Transplant 10 [Suppl 8]:3–7
Lieske JC, Monico CG, Holmes WS, Bergstralh EJ, Slezak JM, Rohlinger AL, Olson JB, Milliner DS (2005) International registry for primary hyperoxaluria. Am J Nephrol 25:290–296
Al-Eisa AA, Samhan M, Naseef M (2004) End-stage renal disease in Kuwaiti children: an 8-year experience. Transplant Proc 36:1788–1791
Kamoun A, Lakhoua R (1996) End-stage renal disease of the Tunisian child: epidemiology, etiologies and outcome. Pediatr Nephrol 10:479–482
Cochat P, Koch Nogueira PC, Mahmoud AM, Jamieson NV, Scheinman JI, Rolland MO (1999) Primary hyperoxaluria in infants: medical, ethical and economic issues. J Pediatr 135:746–750
Millan MT, Berquist WE, So SK, Sarwal MM, Wayman KI, Cox KL, Filler G, Salvatierra O Jr, Esquivel CO (2003) One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: a single-center experience. Transplantation 76:1458–1463
Cochat P, Rolland MO (2005) The primary hyperoxalurias. In: Davison AM, Cameron JS, Grünfeld JP, Ponticelli C, Ritz E, Winearls CG, van Ypersele C (eds) Oxford textbook of clinical nephrology (3rd edn). Oxford University Press, Oxford, pp 2374–2380
Danpure CJ (2005) Molecular etiology of primary hyperoxaluria type 1: new directions for treatment. Am J Nephrol 25:303–310
Daudon M, Jungers P (2004) Clinical value of crystalluria and quantitative morphoconstitutional analysis of urinary calculi. Nephron Physiol 98:31–36
Wong PN, Law ELK, Tong GMW, Mak SK, Lo KY, Wong AKM (2003) Diagnosis of primary hyperoxaluria type 1 by determination of peritoneal dialysate glycolic acid using standard organic-acids analysis method. Perit Dial Int 23:S210–S213
Coulter-Mackie MB, Rumsby G (2004) Genetic heterogeneity in primary hyperoxaluria type 1: impact on diagnosis. Mol Genet Metab 83:38–46
Amoroso A, Pirulli D, Florian F, Puzzer D, Boniotto M, Crovella S, Zezlina S, Spano A, Mazzola G, Savoldi S, Ferrettini C, Berutti S, Petrarulo M (2001) AGXT gene mutations and their influence on clinical heterogeneity of type 1 primary hyperoxaluria. J Am Soc Nephrol 12:2072–2079
Marangella M, Vitale C, Petrarulo M, Tricerri A, Cerelli E, Cadario A, Barbos MP, Linari F (1995) Bony content of oxalate in patients with primary hyperoxaluria or oxalosis-unrelated renal failure. Kidney Int 48:182–187
Danpure CJ, Rumsby G (1995) Enzymological and molecular genetics of primary hyperoxaluria type 1. Consequences for clinical management. In: Khan SR (ed) Calcium oxalate in biological systems. CRC Press, Boca Raton, pp 189–205
Leumann E, Hoppe B (2005) Primary hyperoxaluria type 1: is genotyping clinically helpful? Pediatr Nephrol 20:555–557
Coulter-Mackie MB, Applegarth D, Toone JR, Henderson H (2004) The major allele of the alanine:glyoxylate aminotransferase gene: seven novel mutations causing primary hyperoxaluria. Mol Genet Metab 82:64–68
Danpure CJ (2004) Molecular aetiology of primary hyperoxaluria type 1. Nephron Exp Nephrol 98:e39–e44
Lumb MJ, Danpure CJ (2000) Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease-causing mutations. J Biol Chem 275:36415–36422
Monico CG, Persson M, Ford GC, Rumsby G, Milliner DS (2002) Potential mechanisms of marked hyperoxaluria not due to primary hyperoxaluria I or II. Kidney Int 62:392–400
Nogueira PC, Vuong TS, Bouton O, Maillard A, Marchand M, Rolland MO, Cochat P, Bozon D (2000) Partial deletion of the AGXT gene (EX1_EX7del): a new genotype in hyperoxaluria type 1. Hum Mutat 15:384–385
Rumsby G, Williams E, Coulter-Mackie M (2004) Evaluation of mutation screening as a first line test for the diagnosis of primary hyperoxaluria. Kidney Int 66:959–96323
Basmaison O, Rolland MO, Cochat P, Bozon D (2000) Identification of 5 novel mutations in the AGXT gene. Hum Mutat 15:577
van Woerden CS, Groothoff JW, Wijburg FA, Annink C, Wanders RJ, Waterham HR (2004) Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int 66:746–752
Monico CG, Rossetti S, Olson JB, Milliner DS (2005) Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int 67:1704–1709
Coulter-Mackie MB (2005) Preliminary evidence for ethnic differences in primary hyperoxaluria type 1 genotype. Am J Nephrol 25:264–268
von Schnakenburg C, Hulton SA, Milford DV, Roper HP, Rumsby G (1998) Variable presentation of primary hyperoxaluria type 1 in 2 patients homozygous for a novel combined deletion and insertion mutation in exon 8 of the AGXT gene. Nephron 78:485–488
Leumann E, Hoppe B (2001) The primary hyperoxalurias. J Am Soc Nephrol 12:1986–1993
Milliner DS, Wilson DM, Smith LH (2001) Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int 2001 59:31–36
Marangella M, Petrarulo M, Cosseddu D, Vitale C, Linari F (1992) Oxalate balance studies in patients on hemodialysis for type I primary hyperoxaluria. Am J Kidney Dis 19:546–553
Hoppe B, Kemper MJ, Bokenkamp A, Portale A, Cohn R, Langman C (1999) Plasma calcium oxalate super saturation in children with primary hyperoxaluria and end-stage renal failure. Kidney Int 56:268–274
Yamauchi T, Quillard M, Takahasi S, Nguyen-Khoa M (2001) Oxalate removal by daily dialysis in a patient with primary hyperoxaluria type 1. Nephrol Dial Transplant 16:2407–2411
Scheinman JI, Najarian JS, Mauer SM (1984) Successful strategies for renal transplantation in primary oxalosis. Kidney Int 25:804–811
Behnke B, Kemper MJ, Kruse HP, Müller-Wiefel DE (2001) Bone mineral density in children with primary hyperoxaluria type 1. Nephrol Dial Transplant 16:2236–2239
Cibrik DM, Kaplan B, Arndorfer JA, Meier-Kriesche HU (2002) Renal allograft survival in patients with oxalosis. Transplantation 74:707–710
Jamieson NV (2005) A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): the European PH1 transplant registry experience 1984–2004. Am J Nephrol 25:282–289
Cochat P, Schärer K (1993) Should liver transplantation be performed before advanced renal insufficiency in primary hyperoxaluria type 1? Pediatr Nephrol 7:212–218
Kemper MJ (2005) The role of preemptive liver transplantation in primary hyperoxaluria type 1. Urol Res 33:376–379
Gagnadoux MF, Lacaille F, Niaudet P, Revillon Y, Jouvet P, Jan D, Guest G, Charbit M, Broyer M (2001) Long term results of liver–kidney transplantation in children with primary hyperoxaluria. Pediatr Nephrol 16:946–950
Ellis SR, Hulton SA, McKiernan PJ, de Ville de Goyet J, Kelly DA (2001) Combined liver–kidney transplantation for primary hyperoxaluria in young children. Nephrol Dial Transplant 16:348–354
Sidhu H, Hoppe B, Hesse A, Tenbrock K, Bromme S, Rietschel E, Peck AB (1998) Absence of Oxalobacter formigenes in cystic fibrosis patients: a risk factor for hyperoxaluria. Lancet 352:1026–1029
Chetyrkin SV, Kim D, Belmont JM, Scheinman JI, Hudson BG, Voziyan PA (2004) Pyridoxamine lowers kidney crystals in experimental hyperoxaluria: a potential therapy for primary hyperoxaluria. Kidney Int 67:53–60
Danpure CJ (2005) Primary hyperoxaluria: from gene defects to designer drugs? Nephrol Dial Transplant 20:1525–1529
Koul S, Johnson T, Pramanik S, Koul H (2005) Cellular transfection to deliver alanine-glyoxylate aminotransferase to hepatocytes: a rational gene therapy for primary hyperoxaluria-1 (PH-1). Am J Nephrol 25:176–182
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cochat, P., Liutkus, A., Fargue, S. et al. Primary hyperoxaluria type 1: still challenging!. Pediatr Nephrol 21, 1075–1081 (2006). https://doi.org/10.1007/s00467-006-0124-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-006-0124-4