
Abstract
Background
Variants in genes encoding nuclear pore complex (NPC) proteins are a newly identified cause of paediatric steroid-resistant nephrotic syndrome (SRNS). Recent reports describing NUP93 variants suggest these could be a significant cause of paediatric onset SRNS. We report NUP93 cases in the UK and demonstrate in vivo functional effects of Nup93 depletion in a fly (Drosophila melanogaster) nephrocyte model.
Methods
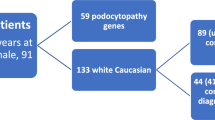
Three hundred thirty-seven paediatric SRNS patients from the National cohort of patients with Nephrotic Syndrome (NephroS) were whole exome and/or whole genome sequenced. Patients were screened for over 70 genes known to be associated with Nephrotic Syndrome (NS). D. melanogaster Nup93 knockdown was achieved by RNA interference using nephrocyte-restricted drivers.
Results
Six novel homozygous and compound heterozygous NUP93 variants were detected in 3 sporadic and 2 familial paediatric onset SRNS characterised histologically by focal segmental glomerulosclerosis (FSGS) and progressing to kidney failure by 12 months from clinical diagnosis.
Silencing of the two orthologs of human NUP93 expressed in D. melanogaster, Nup93-1, and Nup93-2 resulted in significant signal reduction of up to 82% in adult pericardial nephrocytes with concomitant disruption of NPC protein expression. Additionally, nephrocyte morphology was highly abnormal in Nup93-1 and Nup93-2 silenced flies surviving to adulthood.
Conclusion
We expand the spectrum of NUP93 variants detected in paediatric onset SRNS and demonstrate its incidence within a national cohort. Silencing of either D. melanogaster Nup93 ortholog caused a severe nephrocyte phenotype, signaling an important role for the nucleoporin complex in podocyte biology.
Graphical Abstract
A higher resolution version of the Graphical abstract is available as Supplementary information

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nuclear pore complexes (NPCs) are channels composed of evolutionarily conserved proteins called nucleoporins (NUPs), penetrating the nuclear envelope to tightly regulate transport of proteins and RNA between the nucleoplasm and cytoplasm [1]. Multiple copies of about 30 NUPs arranged around the central transport channel form each NPC. In vertebrates this structure is pre-assembled into a core scaffold made of the NUP93/NUP205 complex and two rings formed by the NUP107/NUP160 complex [2]. Additional roles for NUP proteins are emerging, beyond nucleocytoplasmic transport, including transcriptional regulation, DNA damage response and chromosome segregation in mitosis [2]. NUP93 in particular has been linked to ciliary function and cell type specific regulation of gene expression [3, 4]. Depletion of NUP93 impairs correct assembly of the nuclear pore [5, 6].
NPC composition varies among cell types, with tissue-specific transport functions and regulatory roles [7] and mutations previously linked to phenotypes such as atrial fibrillation [8] and leukaemia [9]. In the kidney, NUP93 is expressed in all kidney cell types. As such, NUP93 mutations reduce signal intensity in both glomerular and extra-glomerular cells of kidney cortex [10]. However, in vitro knockdown of NUP93 in human podocytes reduces cell migration, proliferation rate and expression of NUP205, whereas overexpression of SRNS-associated NUP93 variants in NUP93-deleted X.laevis egg extracts lead to disrupted assembly of the NPC [11]. NUP93 mutations also abolish protein–protein interaction with SMAD4 and importin 7 potentially disrupting nucleocytoplasmic trafficking [11]. Equally, RNAi depletion of NUP93 in C.elegans in vivo results in atypical NPC distribution within the nuclear envelope and failure in nuclear exclusion of non-nuclear macromolecules [12]. No in vivo model of NUP93 knockdown has yet been described that disturbs podocyte or glomerular function.
D. melanogaster nephrocytes are podocyte-like and situated pericardially, adjacent to the heart tube. Analogous to mammalian podocytes, insect nephrocytes also form slit diaphragms [13, 14] with evolutionary conservation of many of the slit diaphragm proteins making D.melanogaster an important animal model of human podocyte biology [15,16,17]. Two orthologues of human NUP93, Nup93-1 and Nup93-2, are expressed; while NUP93-2 has widespread tissue expression, NUP93-1 is virtually specific to pericardial nephrocytes [18].
Mutations in 7 nuclear pore associated genes, NUP85, NUP107, NUP133, NUP160, NUP205, XP05 and NUP93 have recently been implicated in paediatric SRNS forming the first kidney-specific example of NPC dysfunction [11, 19, 20]. To date, 15 different NUP93 mutations are reported in 16 SRNS patients with the most common variant, p.(Gly591Val), found in 9/16 cases [11, 21,22,23,24]. We recently described a UK cohort of paediatric SRNS patients, collected via a national UK Renal Registry (RaDaR) [25]. Exome sequencing was performed on the first 187 patients, with an additional 150 patients also whole genome sequenced. All patients were initially screened for mutations in the 76 genes currently known to be associated with NS (Supplementary Table 1).
We were surprised to find a higher-than-expected incidence of novel or very rare variants in NUP93 in 4 unrelated pedigrees within this relatively small paediatric cohort compared with that of other previously-described podocyte gene mutations. As such, we expand the description of NUP93 variants in SRNS, and our experimental results provide the first in vivo evidence that NPC disruption, resulting from NUP93-1 and/or NUP93-2 knockdown in the D. melanogaster nephrocyte, significantly affects glomerular filtration barrier function.
We propose that this in vivo model has the potential to serve as a gold standard in the future for functional assay of model single missense variants in NUP93 to establish likely pathogenicity.
Material and methods
Sequencing
Patient 85 was whole exome sequenced as previously described [25]. Patient 7 tested negative for 24 genes associated with SRNS [26], so was subsequently whole genome sequenced within the WGS500 project (Oxford Genomics Centre at the Wellcome Centre for Human Genetics) [27]. His sister, 7S, was screened at the Bristol Genetics Laboratory to confirm the gene variant (SRNS gene panel, www.nbt.nhs.uk/severn-pathology/pathology-services/bristol-genetics-laboratory-bgl). Patients S013282 and S013682 were whole genome sequenced at the NIHR BioResource (Cambridge, UK) [28]. Sequencing was performed on an Illumina platform with mean coverage of 35X. Variant calling and annotation were performed using the King’s College London BRC Genomics [25] and NIHR BioResource [28] pipelines. All detected variants were filtered and classified as described previously [25, 29] and the description of each step is described in the Supplemental Data.
Homozygosity mapping [30] was performed for patient S013682. All homozygous variants found within the coding sequence and the detected stretches of homozygosity were analysed. Variants with MAF > 0.01 or seen as homozygotes in gnomAD database or an in-house control database or affecting a non-conserved amino acid were filtered out. Synonymous variants were ignored since they are less likely to affect the protein.
Animal model—NUP93 knock-down in Drosophila
Flies were raised on a standard cornmeal-yeast diet at 25 °C under a 12:12 h light:dark cycle. Knockdown was achieved by RNA interference using the following fly strains: UAS-NUP 93–2 RNAi (Bloomington stock centre, number 51758,); UAS-NUP93-1 (Bloomington stock centre, number 34090, both Harvard TRiP lines containing a targeting hairpin insertion in the third chromosome [31], DotGal4[32] and dKlf15Gal4 [33].
Staining of adult Drosophila nephrocytes
For staining, 4–9 day old flies were dissected as previously described [34]. In brief, dissected hearts and nephrocytes were fixed in 3.8% formaldehyde in PBS for 20 min, washed 3 × with PBS and then incubated in wheat germ agglutinin (conjugated to Alexa 594 fluorochrome at 1 ug/ml) and Alexa-Fluor-488 conjugated Phalloidin (1/250) to stain F-actin. To stain the NPC, samples were incubated in primary antibody (Mab414-Abcam) and Hoechst to visualise DNA followed by incubation with relevant secondary antibody (Invitrogen, UK). Images were taken using a Zeiss Axiolab widefield LED fluorescent microscope with water dipping × 10 or × 40 objectives. Micrographs were collected using Micromanager and contrast-enhanced using Photoshop, all images being treated equally.
Statistics
One-way ANOVA followed by Tukey’s post hoc HSD test.
Results
Four patients from the UK paediatric cohort of NS were found to have rare, highly conserved and predicted novel possibly pathogenic variants in the NUP93 gene. One patient was found to have a novel variant but of unknown significance (VUS) (Table 1).
Two heterozygous NUP93 variants were detected in sporadic patient 85. Mother was heterozygous for p.(Leu695Ser); paternal DNA was not available. Both affected amino acids are highly conserved, and in silico tools predicted deleteriousness.
Siblings 7 and 7S shared a novel, homozygous missense p.(Ala475Thr) variant; parents were consanguineous. This variant affects a highly conserved alanine and was predicted to be deleterious.
A homozygous novel p.(Lys637Glu) variant was detected in a second consanguineous case, S013682. Parents were heterozygous for the same variant as was an unaffected maternal uncle. This variant affects a highly conserved amino acid, and the substitution is predicted to be deleterious in silico by 3 out of 5 of the tools used. Homozygosity mapping was performed for additional confirmation. This was able to verify whether the variant falls within a homozygosity run and to test whether there are any alternative variants of interest in this patient. Only 4 homozygous variants were left after the filtering, with p.(Lys637Glu) being the strongest candidate to be associated with the phenotype in this patient (Supplemental Fig. 3 and Table 2).
Two heterozygous variants in trans, inherited from each nonconsanguineous parent, were detected in S013282. The first variant, c.1538-6A > G, located within the consensus sequence in intron 13 was predicted to create an alternative/new acceptor site (Supplemental Fig. 1). The second variant, p.(His491Gln) was predicted deleterious, affecting a highly conserved amino acid. Additionally, this variant was predicted to create a new donor and a new acceptor site (Supplemental Fig. 2).
Four of the NUP93 variants detected are not present in gnomAD and the remaining 2 are rare with a very low minor allele frequency (MAF) at 0.00003536 (counts:10/0/282796) and 0.000004196 (counts:1/0/238330) and not reported as homozygotes [35]. All 6 NUP93 variants are located in the second part of the gene, similar to the previously described mutations [10, 11, 21,22,23], and are within the 2 α-helical domains of Nup93 (Fig. 1). Location of the variants on the predicted human 3D NUP93 structure (AlphaFold) [36] as well as the DynaMut [37] predictions of protein stability change upon the detected amino acid substitutions can be found in Supplemental Figs. 4–6 and Supplemental Table 3.

NUP93 variants A. Exon structure of human NUP93 cDNA with identified variants. In red: variants found in the UK SRNS cohort, in black: mutations previously described in the literature [10, 11, 21,22,23,24, 39, 40]. NUP93 domain structure created with Prosite MyDomains (https://prosite.expasy.org/cgi-bin/prosite/mydomains) [51]. B. NUP93 protein domain structure with variant positions indicated. Tr, transmembrane; C, conservation across evolution of altered amino acids for the 5 missense variants. Alamut Visual version 2.15 (SOPHiA GENETICS, Lausanne, Switzerland)
Phenotypes are listed in Table 2 and compare with previously-reported cases [10, 11, 21,22,23,24]. All had multi-drug resistant focal segmental glomerulosclerosis (FSGS), progressing rapidly to CKD stage 5 within 2–12 months and required kidney transplant. Atypically for monogenic SRNS, post-transplant disease recurrence occurred within 24 h in 1/5 cases, S013682, responding to plasmapheresis (PE) daily for 2 weeks then × 2 weekly for 4 weeks. At 13 years post-transplant, graft function is good with no evidence of proteinuria. Patient 7 passed away 2 years after the transplant from respiratory failure. Four out of 5 patients had microscopic haematuria pre-transplantation as described previously in SRNS caused by NUP93 mutations [11].
Histology
All 5 patients had FSGS on biopsy. Patient 7 (Fig. 2) also had features of collapsing glomerulopathy, and together with his sibling 7S, tubular dilatation. Patients 7S, 85, S013682 and S013282 had interstitial fibrosis and tubular atrophy consistent with previous reports of NUP93 patients [10, 11]. Full histological description is provided in Supplementary Data.

Electron microscopy images of a kidney biopsy sample from patient 7 and 7S. Left panel shows electron micrographs of kidney biopsy sample from patient 7. There is gross foot process fusion with only a few areas showing intact foot processes. Otherwise, the glomeruli are of normal architecture without evidence of electron dense deposit. There is no significant mesangial thickening and GBMs appear of normal thickness. Right panel shows electron micrographs of kidney biopsy sample from patient 7S. The glomerulus is partly contracted. Endothelial hyperplasia and subendothelial oedema are observed (dashed arrow). In some areas of the GBM there appears to be some lamination and thickening of the basement membrane together with a basketweave-like appearance that is on the epithelial (outside) aspect of the membrane (solid arrow)
D. melanogaster model
In view of these findings, and the similarities between human podocytes and D.melanogaster nephrocytes, we developed an animal model to investigate the effect of Nup93 knockdown.
The impact of silencing Nup93 in nephrocytes is shown in Fig. 3. Silencing Nup93-1 or Nup93-2 using nephrocyte-restricted drivers led to a significant reduction in adult pericardial nephrocyte numbers (Fig. 3A and B). Using the DotGal4 driver, nephrocyte numbers were reduced by 70% (Nup93-1 silencing; P < 0.001) or 82% (Nup93-2 silencing; P < 0.001). Similarly, using the dKlf15Gal4 driver, nephrocyte numbers were reduced 50% and 36% (Nup93-1 and Nup93-2, respectively; P < 0.001 for both compared to controls). Antibody staining against the nuclear pore complex proteins was robust in wild type and parent lines but grossly disrupted, with loss of nuclear membrane localisation, in Nup93-1 or Nup93-2 silenced nephrocytes. In addition, nephrocytes in flies surviving to adulthood showed enlarged and granular morphology in Nup93-1 and Nup93-2 silenced flies (Fig. 3C).

NUP93 is crucial for Drosophila pericardial nephrocyte development. (A) Micrographs of the adult Drosophila heart stained with wheat germ agglutinin (WGA, red) and phalloidin (green). The heart tube (HT) of wild-type flies (2 individuals are shown) is flanked by kidney-like nephrocytes (arrows) which preferentially bind WGA. In contrast, fewer nephrocytes are seen in adults when the expression of either NUP93-1 or NUP93-2 is silenced by RNAi using a nephrocyte-restricted driver Dot-Gal4 (arrowheads). Scale bars = 100 µm. 7. (B) Quantified data indicate a significant reduction in nephrocyte numbers in flies where NUP93-1 or NUP93-2 were silenced using either DotGal4 or dKlf15Gal4 drivers, compared to wild type (w1118) or parent lines (DotGal4; dKlf15Gal4; UAS-NUP93-1 RNAi or UAS-NUP92-2 RNAi). n = 22–35 different flies for each genotype. *P < 0.001 compared to w1118 control line. (C) Micrographs show nephrocytes stained with anti-nucleopore protein antibody (mAB 414, red), wheat germ agglutinin (WGA, green; cell membranes) and Hoechst (blue, nucleic acids). Nephrocytes in wild-type flies (in this instance, the driver line outcrossed to w1118) show distinct perinuclear staining of nucleopore proteins (arrow), whereas the nucleus and perinuclear staining for nucleopore proteins is severely disrupted in nephrocytes when NUP93-1 was silenced (arrowhead, dKlf15Gal4 > NUP93-1 RNAi). Scale bar = 20 µm
Wild type nephrocytes were inflated with a convex surface on EM, with slit-like structures highly similar to the podocyte filtration system [13] (Fig. 4). The cytoplasmic side of these openings into the cell interior are dense where they approximate to the external membrane. These slits lead into a sub-membranous labyrinth of interconnected spaces with an external well-formed basement membrane (Fig. 4, WT control higher magnification).

Electron microscopy of NUP93 mutants and wild type Drosophila nephrocytes. Top panel shows low power sections, bottom panel shows higher power sections. NUP93-1: A collapsed cellular profile with indented margins and large vacuoles abundant in the cytoplasm. Basement membrane is intact with very few fenestrations leading into large interconnecting sub-membranous spaces containing membrane bound inclusions. NUP93-2: An over-inflated cellular profile with large vacuoles abundant in the cytoplasm. Basement membrane is intact with fewer fenestrations than control leading into a well-defined narrow sub-membranous labyrinth. Wild type control: An inflated cellular profile with large vacuoles abundant in the cytoplasm. Basement membrane is intact with abundant fenestrations leading into well-defined narrow sub-membranous labyrinths
Mutant nephrocytes had an increased number of dents or concave surface regions at low magnification (Fig. 4Nup93-1). At times, cells were swollen (Fig. 4Nup93-2) with the cell surface only partially covered in slits leading to a disrupted sub-membranous labyrinth (Fig. 4NUP93-1 and NUP93-2 higher magnification). The external basement membrane appears similar to the control.
Discussion
To date, mutations in 7 nuclear pore genes, NUP85, NUP107, NUP133, NUP160, NUP205, XP05, and NUP93 have been implicated in SRNS [11, 19, 20], with 16 patients (15 families) described with NUP93. Here, we report a further 6 novel and previously undescribed NUP93 variants in 3 sporadic (85, S013282 and S013682) and 2 familial (7 and 7S) paediatric-onset SRNS cases. Of note, the most common previously-identified p.(Gly591Val) NUP93 mutation (9/16 cases) was absent in our cohort suggestive of a founder effect in those patients [11, 21]. We used stringent phenotyping as well as bioinformatic approaches for genetic data analysis and also used these to support in silico the possibly pathogenic role in the detected variants (variants were considered as possibly disease-causing when they were novel or very rare and predicted to affect protein [25]). While we provide compelling computational evidence for their potential pathogenicity and the fact that they are likely causal in at least 4 of these individuals’ disease (especially in the absence of any other candidate genes), each variant would require further functional confirmation either in a cellular or animal model or identified in other unrelated SRNS patients.
However, NUP93 mutations have only recently been associated with SRNS, and as such only a small number of pathogenic mutations in limited kindreds have been identified to date. As a result, and in common with other new genes in rare diseases, mutational hotspots or critical regions have yet to be identified. This lack of gene data and absence of functional proof in a cellular or animal model meant that despite strong computational evidence for likely pathogenicity and robust genotype–phenotype correlation, for novel or very rare variants current ACM guidelines are often not met [38]. Consequently, without functional assay and/or identification in other (unrelated) SRNS patients, not only the NUP93 variants described in this paper but the majority previously published, would be classified as variants of unknown significance (VUS). This leads to the important discussion about how best to apply guidelines in rare disease bearing in mind the rapid and increasing application of next generation sequencing as a diagnostic tool in kidney medicine. Fortunately, interpretation using a composite picture made up of phenotype, genotype and results of conventional tests such as kidney biopsy can refine the likelihood of a lab report of a “VUS” as more likely pathogenic, pending results of functional assay or more gene data.
Our data is consistent with previous reports [10, 11, 21,22,23,24, 39, 40], supporting NUP93 mutations as a cause of paediatric onset SRNS. Familial cases had homozygous changes p.(Ala475Thr, 7 and 7 s) and p.(Lys637Glu, S013682) and were diagnosed in the first 2 years of life with rapid onset kidney failure. Sporadic patients 85 and S013282 were diagnosed at age 6 and 8 respectively with later-onset SRNS but correspondingly dramatic decline in eGFR within 2–12 months. All 5 had FSGS, tubular atrophy and dilatation, hyaline casts on kidney biopsy and haematuria consistent with previous reports [10, 11]. Unusually for multidrug resistant monogenic disease [41], S013682 developed post-transplant disease recurrence responding to plasma exchange (PEX). Interestingly, post-transplant disease recurrence responding to PEX has been described previously in a case of SRNS caused by compound heterozygous NUP93 mutation (p.Gly591Val + p.Leu639Pro) [21, 22]. In contrast to S013682 successfully treated with PEX alone, the case became PEX dependent and required rituximab [22]. Post-transplant recurrence is rare in monogenic SRNS and is generally mediated by antibodies, most descriptions being of anti-nephrin antibodies in Finnish type congenital nephrotic syndrome [42]. Equally, there are links between nucleoporins and innate immunity raising the possibility of an as yet unidentified mechanism. The other possibility is that the NUP93 variant in this patient is benign, emphasising the need for improved prediction models.
To this end we confirm that nephrocyte-specific knockdown of Nup93 in vivo is deleterious, supporting a direct effect of NUP93 loss of function mutations on podocyte function and therefore a pathogenic role in SRNS. Since novel mutations without supporting functional data tend to be classified as “VUS”, our Drosophila in vivo model could be modified to assay NUP93 mutations to determine pathogenicity in this and other SRNS cohorts.
The need for better experimental models is illustrated well by our patient with post-transplant recurrence. The location of p.(Lys637Glu) is within a region described for other NUP93 mutations. In addition, this novel variant is absent in healthy ethnically-matched controls and commonly used bioinformatics analysis determined a deleterious score for this amino acid substitution. However, the evolutionary model of variant effect (EVE) recently published in Nature [43], predicts the amino acid substitution to be benign. Alleles were inherited in trans, i.e., one from each unaffected heterozygous parent. Homozygosity mapping analysis further supported the variant being the strongest candidate in this patient. Functional work such as in our fly model, would be required at this stage to confirm pathogenicity and reclassify this variant from “uncertain significance.” Post-transplant recurrence observed in patient S013682 remains unexplained and the relationship between the genetic variant and recurrence remains to be seen. However, this is the second case of post-transplant disease recurrence associated with a NUP93 mutation. Sandokji et al. recently reported a 5-year-old girl presenting with SRNS, cardiomyopathy and developmental delay with autistic features. After kidney transplant, she had self-limiting proteinuria which resolved within a week [24]
Nucleoporins are known to have a role in the immune system. Although there is no corresponding NUP93 mouse model, NUP210 knockout mice develop peripheral T-cell alterations [44] while NUP96 + / − animals present selective alterations of the immune system [45]. Furthermore, NUP93 interacts with SMAD4, a signaling protein, and NUP93 mutations have been shown to abrogate SMAD activity [11]. SMAD4 has a critical role in T-cell function and is required in T-cell-mediated autoimmunity and tumour rejection [46]. NUP93 is also known to regulate antiviral innate immune responses [47] and is expressed in peripheral blood mononuclear cells as well as in the kidney [10]. This apparent link between the immune system and other nucleoporins, together with NUP93 expression in mononuclear cells, supports a relationship with immune function as does the detection of anti-NUP210 and -NUP62 antibodies in primary biliary cirrhosis, systemic lupus erythematosus, autoimmune myositis and rheumatic diseases [48]. Nevertheless, a link with risk of disease recurrence post-transplant remains unproven.
In common with other nucleoporins [20] the NUP93 phenotype can either be a kidney-specific disease or syndromic with neurodevelopmental features and cardiac anomalies [24]. NUP93 is highly expressed in the human brain and cerebellum and NUP93 mutations were recently linked to autosomal recessive congenital ataxia [49]. While none of our cases had ataxia, autistic spectrum disorder was present in 1 of our patients, S013282, and more non-specific features of neurodevelopmental delay in the other patients as might be expected. This confirms the previously noted association and affirms that SRNS cases with NUP93 mutations may also have neurological involvement.
NUP93 is expressed in all human kidney cell types [10]. Cellular knockdown of NUP93 in vitro has been shown to be significantly important in cell functions such as nuclear pore assembly, cell migration, and interaction with signaling cascading mediated by other NUP’s, SMAD4 and importin 7 [11, 50]. Since D. melanogaster and humans share conserved roles in a number of cellular processes, we opted to use the fly’s equivalent of podocytes, the nephrocyte, to investigate a role for NUP93 in SRNS.
We created the first in vivo nephrocyte specific knockdown of Nup93. Examination of our experimental model indicated that nephrocyte development requires the expression of both human NUP93 orthologues, ubiquitously expressed Nup93-1 and nephrocyte-specific Nup93-2. Silencing expression using either of 2 well-defined, nephrocyte-restricted drivers led to a reduced number of nephrocytes with surviving cells demonstrating highly abnormal morphology. Neither the Nup93-1 nor the Nup93-2 RNAi lines are predicted to have off-target effects, and silencing of either independently caused a severe phenotype, indicating a lack of functional compensation. These findings support using a fly model as a functional assay to determine the pathogenicity of mutations detected in human disease.
In summary, we detected 6 new NUP93 variants in autosomal recessive paediatric-onset SRNS expanding the variation spectrum and confirming that NUP93 is associated with a phenotype of multi-drug resistant, rapidly progressive SRNS. Phenotypic expansion to include neurodevelopmental features if syndromic disease is present is also confirmed. The potential for NUP93 mutations as causal in podocytopathy is supported mechanistically with the finding that silencing either Nup93-1 or Nup93-2 leads to a severe and non-redundant disruption of D. melanogaster nephrocyte development and function. Our data underpins the difficulties in confirming mutations as pathogenic in rare diseases with recently described gene associations and with only a few cases described, despite strong correlation between phenotype and genotype. While it is recognised that this is difficult to implement in the clinic, the rapid generation of data using next generation sequencing and the current difficulties in interpreting “pertinent” from “incidental” supports the need for functional assay of mutations such as in the fly kidney model presented here.
Data availability
All data and materials are stored according to Ethical approvals at Bristol Renal laboratories.
References
Kabachinski G, Schwartz TU (2015) The nuclear pore complex–structure and function at a glance. J Cell Sci 128:423–429. https://doi.org/10.1242/jcs.083246
Ibarra A, Hetzer MW (2015) Nuclear pore proteins and the control of genome functions. Genes Dev 29:337–349. https://doi.org/10.1101/gad.256495.114
Del Viso F, Huang F, Myers J, Chalfant M, Zhang Y, Reza N, Bewersdorf J, Lusk CP, Khokha MK (2016) Congenital Heart Disease Genetics Uncovers Context-Dependent Organization and Function of Nucleoporins at Cilia. Dev Cell 38:478–492. https://doi.org/10.1016/j.devcel.2016.08.002
Ibarra A, Benner C, Tyagi S, Cool J, Hetzer MW (2016) Nucleoporin-mediated regulation of cell identity genes. Genes Dev 30:2253–2258. https://doi.org/10.1101/gad.287417.116
Grandi P, Dang T, Pane N, Shevchenko A, Mann M, Forbes D, Hurt E (1997) Nup93, a vertebrate homologue of yeast Nic96p, forms a complex with a novel 205-kDa protein and is required for correct nuclear pore assembly. Mol Biol Cell 8:2017–2038. https://doi.org/10.1091/mbc.8.10.2017
Sachdev R, Sieverding C, Flotenmeyer M, Antonin W (2012) The C-terminal domain of Nup93 is essential for assembly of the structural backbone of nuclear pore complexes. Mol Biol Cell 23:740–749. https://doi.org/10.1091/mbc.E11-09-0761
Capelson M, Hetzer MW (2009) The role of nuclear pores in gene regulation, development and disease. EMBO Rep 10:697–705. https://doi.org/10.1038/embor.2009.147
Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R, Wang L, Chen Q, Wang QK (2008) Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135:1017–1027. https://doi.org/10.1016/j.cell.2008.10.022
Xu S, Powers MA (2009) Nuclear pore proteins and cancer. Semin Cell Dev Biol 20:620–630. https://doi.org/10.1016/j.semcdb.2009.03.003
Hashimoto T, Harita Y, Takizawa K, Urae S, Ishizuka K, Miura K, Horita S, Ogino D, Tamiya G, Ishida H, Mitsui T, Hayasaka K, Hattori M (2019) In Vivo Expression of NUP93 and Its Alteration by NUP93 Mutations Causing Focal Segmental Glomerulosclerosis. Kidney Int Rep 4:1312–1322. https://doi.org/10.1016/j.ekir.2019.05.1157
Braun DA, Sadowski CE, Kohl S, Lovric S, Astrinidis SA, Pabst WL, Gee HY, Ashraf S, Lawson JA, Shril S, Airik M, Tan W, Schapiro D, Rao J, Choi WI, Hermle T, Kemper MJ, Pohl M, Ozaltin F, Konrad M, Bogdanovic R, Buscher R, Helmchen U, Serdaroglu E, Lifton RP, Antonin W, Hildebrandt F (2016) Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 48:457–465. https://doi.org/10.1038/ng.3512
Galy V, Mattaj IW, Askjaer P (2003) Caenorhabditis elegans nucleoporins Nup93 and Nup205 determine the limit of nuclear pore complex size exclusion in vivo. Mol Biol Cell 14:5104–5115. https://doi.org/10.1091/mbc.e03-04-0237
Weavers H, Prieto-Sanchez S, Grawe F, Garcia-Lopez A, Artero R, Wilsch-Brauninger M, Ruiz-Gomez M, Skaer H, Denholm B (2009) The insect nephrocyte is a podocyte-like cell with a filtration slit diaphragm. Nature 457:322–326. https://doi.org/10.1038/nature07526
Zhuang S, Shao H, Guo F, Trimble R, Pearce E, Abmayr SM (2009) Sns and Kirre, the Drosophila orthologs of Nephrin and Neph1, direct adhesion, fusion and formation of a slit diaphragm-like structure in insect nephrocytes. Development 136:2335–2344. https://doi.org/10.1242/dev.031609
Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V, Zhou W, Lovric S, Fang H, Nettleton M, Zhu JY, Hoefele J, Weber LT, Podracka L, Boor A, Fehrenbach H, Innis JW, Washburn J, Levy S, Lifton RP, Otto EA, Han Z, Hildebrandt F (2015) KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest 125:2375–2384. https://doi.org/10.1172/JCI79504
Helmstadter M, Luthy K, Godel M, Simons M, Ashish ND, Rensing SA, Fischbach KF, Huber TB (2012) Functional study of mammalian Neph proteins in Drosophila melanogaster. PLoS One 7:e40300. https://doi.org/10.1371/journal.pone.0040300
Na J, Sweetwyne MT, Park AS, Susztak K, Cagan RL (2015) Diet-Induced Podocyte Dysfunction in Drosophila and Mammals. Cell Rep 12:636–647. https://doi.org/10.1016/j.celrep.2015.06.056
Chintapalli VR, Wang J, Dow JA (2007) Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39:715–720. https://doi.org/10.1038/ng2049
Miyake N, Tsukaguchi H, Koshimizu E, Shono A, Matsunaga S, Shiina M, Mimura Y, Imamura S, Hirose T, Okudela K, Nozu K, Akioka Y, Hattori M, Yoshikawa N, Kitamura A, Cheong HI, Kagami S, Yamashita M, Fujita A, Miyatake S, Tsurusaki Y, Nakashima M, Saitsu H, Ohashi K, Imamoto N, Ryo A, Ogata K, Iijima K, Matsumoto N (2015) Biallelic Mutations in Nuclear Pore Complex Subunit NUP107 Cause Early-Childhood-Onset Steroid-Resistant Nephrotic Syndrome. Am J Hum Genet 97:555–566. https://doi.org/10.1016/j.ajhg.2015.08.013
Braun DA, Lovric S, Schapiro D, Schneider R, Marquez J, Asif M, Hussain MS, Daga A, Widmeier E, Rao J, Ashraf S, Tan W, Lusk CP, Kolb A, Jobst-Schwan T, Schmidt JM, Hoogstraten CA, Eddy K, Kitzler TM, Shril S, Moawia A, Schrage K, Khayyat AIA, Lawson JA, Gee HY, Warejko JK, Hermle T, Majmundar AJ, Hugo H, Budde B, Motameny S, Altmuller J, Noegel AA, Fathy HM, Gale DP, Waseem SS, Khan A, Kerecuk L, Hashmi S, Mohebbi N, Ettenger R, Serdaroglu E, Alhasan KA, Hashem M, Goncalves S, Ariceta G, Ubetagoyena M, Antonin W, Baig SM, Alkuraya FS, Shen Q, Xu H, Antignac C, Lifton RP, Mane S, Nurnberg P, Khokha MK, Hildebrandt F (2018) Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J Clin Invest 128:4313–4328. https://doi.org/10.1172/JCI98688
Bezdicka M, Stolbova S, Seeman T, Cinek O, Malina M, Simankova N, Pruhova S, Zieg J (2018) Genetic diagnosis of steroid-resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: a high proportion of causative variants in NUP93. Pediatr Nephrol 33:1347–1363. https://doi.org/10.1007/s00467-018-3950-2
Seeman T, Vondrak K (2018) First Report of Recurrent Nephrotic Syndrome After Kidney Transplantation in a Patient With NUP93 Gene Mutations: A Case Report. Transplant Proc 50:3954–3956. https://doi.org/10.1016/j.transproceed.2018.07.010
Rossanti R, Shono A, Miura K, Hattori M, Yamamura T, Nakanishi K, Minamikawa S, Fujimura J, Horinouchi T, Nagano C, Sakakibara N, Kaito H, Nagase H, Morisada N, Asanuma K, Matsuo M, Nozu K, Iijima K (2019) Molecular assay for an intronic variant in NUP93 that causes steroid resistant nephrotic syndrome. J Hum Genet 64:673–679. https://doi.org/10.1038/s10038-019-0606-4
Sandokji I, Marquez J, Ji W, Zerillo CA, Konstantino M, Lakhani SA, Khokha MK, Warejko JK (2019) Identification of novel mutations and phenotype in the steroid resistant nephrotic syndrome gene NUP93: a case report. BMC Nephrol 20:271. https://doi.org/10.1186/s12882-019-1458-z
Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, Nabhan MM, Kerecuk L, Hegde S, Hughes D, Marks S, Feather S, Jones C, Webb NJ, Ognjanovic M, Christian M, Gilbert RD, Sinha MD, Lord GM, Simpson M, Koziell AB, Welsh GI, Saleem MA (2017) Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int 91:937–947. https://doi.org/10.1016/j.kint.2016.10.013
McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, Sinha MD, Webb NJ, Christian M, Williams MM, Marks S, Koziell A, Welsh GI, Saleem MA; RADAR the UK SRNS Study Group (2013) Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 8:637–648. https://doi.org/10.2215/CJN.07200712
Taylor JC, Martin HC, Lise S, Broxholme J, Cazier JB, Rimmer A, Kanapin A, Lunter G, Fiddy S, Allan C, Aricescu AR, Attar M, Babbs C, Becq J, Beeson D, Bento C, Bignell P, Blair E, Buckle VJ, Bull K, Cais O, Cario H, Chapel H, Copley RR, Cornall R, Craft J, Dahan K, Davenport EE, Dendrou C, Devuyst O, Fenwick AL, Flint J, Fugger L, Gilbert RD, Goriely A, Green A, Greger IH, Grocock R, Gruszczyk AV, Hastings R, Hatton E, Higgs D, Hill A, Holmes C, Howard M, Hughes L, Humburg P, Johnson D, Karpe F, Kingsbury Z, Kini U, Knight JC, Krohn J, Lamble S, Langman C, Lonie L, Luck J, McCarthy D, McGowan SJ, McMullin MF, Miller KA, Murray L, Nemeth AH, Nesbit MA, Nutt D, Ormondroyd E, Oturai AB, Pagnamenta A, Patel SY, Percy M, Petousi N, Piazza P, Piret SE, Polanco-Echeverry G, Popitsch N, Powrie F, Pugh C, Quek L, Robbins PA, Robson K, Russo A, Sahgal N, van Schouwenburg PA, Schuh A, Silverman E, Simmons A, Sorensen PS, Sweeney E, Taylor J, Thakker RV, Tomlinson I, Trebes A, Twigg SR, Uhlig HH, Vyas P, Vyse T, Wall SA, Watkins H, Whyte MP, Witty L, Wright B, Yau C, Buck D, Humphray S, Ratcliffe PJ, Bell JI, Wilkie AO, Bentley D, Donnelly P, McVean G (2015) Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 47:717–726. https://doi.org/10.1038/ng.3304
Turro E, Astle WJ, Megy K, Graf S, Greene D, Shamardina O, Allen HL, Sanchis-Juan A, Frontini M, Thys C, Stephens J, Mapeta R, Burren OS, Downes K, Haimel M, Tuna S, Deevi SVV, Aitman TJ, Bennett DL, Calleja P, Carss K, Caulfield MJ, Chinnery PF, Dixon PH, Gale DP, James R, Koziell A, Laffan MA, Levine AP, Maher ER, Markus HS, Morales J, Morrell NW, Mumford AD, Ormondroyd E, Rankin S, Rendon A, Richardson S, Roberts I, Roy NBA, Saleem MA, Smith KGC, Stark H, Tan RYY, Themistocleous AC, Thrasher AJ, Watkins H, Webster AR, Wilkins MR, Williamson C, Whitworth J, Humphray S, Bentley DR, BioResource N, for the GP, Kingston N, Walker N, Bradley JR, Ashford S, Penkett CJ, Freson K, Stirrups KE, Raymond FL, Ouwehand WH (2020) Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583:96–102. https://doi.org/10.1038/s41586-020-2434-2
Bierzynska A, Soderquest K, Dean P, Colby E, Rollason R, Jones C, Inward CD, McCarthy HJ, Simpson MA, Lord GM, Williams M, Welsh GI, Koziell AB, Saleem MA, NephroS; UK study of Nephrotic Syndrome (2017) MAGI2 Mutations Cause Congenital Nephrotic Syndrome. J Am Soc Nephrol 28:1614–1621. https://doi.org/10.1681/ASN.2016040387
Seelow D, Schuelke M, Hildebrandt F, Nurnberg P (2009) HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Res 37:W593–W599. https://doi.org/10.1093/nar/gkp369
Flockhart IT, Booker M, Hu Y, McElvany B, Gilly Q, Mathey-Prevot B, Perrimon N, Mohr SE (2012) FlyRNAi.org--the database of the Drosophila RNAi screening center: 2012 update. Nucleic Acids Res 40:D715–D719. https://doi.org/10.1093/nar/gkr953
Kimbrell DA, Hice C, Bolduc C, Kleinhesselink K, Beckingham K (2002) The Dorothy enhancer has Tinman binding sites and drives hopscotch-induced tumor formation. Genesis 34:23–28. https://doi.org/10.1002/gene.10134
Ivy JR, Drechsler M, Catterson JH, Bodmer R, Ocorr K, Paululat A, Hartley PS (2015) Klf15 is critical for the development and differentiation of Drosophila Nephrocytes. PLoS One 10:e0134620. https://doi.org/10.1371/journal.pone.0134620
Catterson JH, Heck MM, Hartley PS (2013) Fermitins, the orthologs of mammalian Kindlins, regulate the development of a functional cardiac syncytium in Drosophila melanogaster. PLoS One 8:e62958. https://doi.org/10.1371/journal.pone.0062958
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database C, Neale BM, Daly MJ, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Rodrigues CH, Pires DE, Ascher DB (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 46:W350–W355. https://doi.org/10.1093/nar/gky300
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, Lovric S, Ashraf S, Rao J, Hermle T, Jobst-Schwan T, Widmeier E, Majmundar AJ, Schneider R, Gee HY, Schmidt JM, Vivante A, van der Ven AT, Ityel H, Chen J, Sadowski CE, Kohl S, Pabst WL, Nakayama M, Somers MJG, Rodig NM, Daouk G, Baum M, Stein DR, Ferguson MA, Traum AZ, Soliman NA, Kari JA, El Desoky S, Fathy H, Zenker M, Bakkaloglu SA, Muller D, Noyan A, Ozaltin F, Cadnapaphornchai MA, Hashmi S, Hopcian J, Kopp JB, Benador N, Bockenhauer D, Bogdanovic R, Stajic N, Chernin G, Ettenger R, Fehrenbach H, Kemper M, Munarriz RL, Podracka L, Buscher R, Serdaroglu E, Tasic V, Mane S, Lifton RP, Braun DA, Hildebrandt F (2018) Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin J Am Soc Nephrol 13:53–62. https://doi.org/10.2215/CJN.04120417
Zhao B, Chen JY, Liao YB, Li YF, Jiang XM, Bi X, Yang MF, Li L, Cui JJ (2021) Steroid-resistant nephrotic syndrome in infants caused by a novel compound heterozygous mutation of the NUP93: A CARE case report. Medicine (Baltimore) 100:e24627. https://doi.org/10.1097/MD.0000000000024627
Bierzynska A, Saleem MA (2018) Deriving and understanding the risk of post-transplant recurrence of nephrotic syndrome in the light of current molecular and genetic advances. Pediatr Nephrol 33:2027–2035. https://doi.org/10.1007/s00467-017-3793-2
Patrakka J, Ruotsalainen V, Reponen P, Qvist E, Laine J, Holmberg C, Tryggvason K, Jalanko H (2002) Recurrence of nephrotic syndrome in kidney grafts of patients with congenital nephrotic syndrome of the Finnish type: role of nephrin. Transplantation 73:394–403. https://doi.org/10.1097/00007890-200202150-00013
Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, Gal Y, Marks DS (2021) Disease variant prediction with deep generative models of evolutionary data. Nature 599:91–95. https://doi.org/10.1038/s41586-021-04043-8
van Nieuwenhuijze A, Burton O, Lemaitre P, Denton AE, Cascalho A, Goodchild RE, Malengier-Devlies B, Cauwe B, Linterman MA, Humblet-Baron S, Liston A (2018) Mice Deficient in Nucleoporin Nup210 Develop Peripheral T Cell Alterations. Front Immunol 9:2234. https://doi.org/10.3389/fimmu.2018.02234
Faria AM, Levay A, Wang Y, Kamphorst AO, Rosa ML, Nussenzveig DR, Balkan W, Chook YM, Levy DE, Fontoura BM (2006) The nucleoporin Nup96 is required for proper expression of interferon-regulated proteins and functions. Immunity 24:295–304. https://doi.org/10.1016/j.immuni.2006.01.014
Gu AD, Zhang S, Wang Y, Xiong H, Curtis TA, Wan YY (2015) A critical role for transcription factor Smad4 in T cell function that is independent of transforming growth factor beta receptor signaling. Immunity 42:68–79. https://doi.org/10.1016/j.immuni.2014.12.019
Monwan W, Kawasaki T, Hasan MZ, Ori D, Kawai T (2020) Identification of nucleoporin 93 (Nup93) that mediates antiviral innate immune responses. Biochem Biophys Res Commun 521:1077–1082. https://doi.org/10.1016/j.bbrc.2019.11.035
Nofrini V, Di Giacomo D, Mecucci C (2016) Nucleoporin genes in human diseases. Eur J Hum Genet 24:1388–1395. https://doi.org/10.1038/ejhg.2016.25
Zanni G, De Magistris P, Nardella M, Bellacchio E, Barresi S, Sferra A, Ciolfi A, Motta M, Lue H, Moreno-Andres D, Tartaglia M, Bertini E, Antonin W (2019) Biallelic Variants in the Nuclear Pore Complex Protein NUP93 Are Associated with Non-progressive Congenital Ataxia. Cerebellum 18:422–432. https://doi.org/10.1007/s12311-019-1010-5
Chen X, Xu L (2010) Specific nucleoporin requirement for Smad nuclear translocation. Mol Cell Biol 30:4022–4034. https://doi.org/10.1128/MCB.00124-10
Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJ (2008) The 20 years of PROSITE. Nucleic Acids Res 36 (Database issue):D245-D249. https://doi.org/10.1093/nar/gkm977
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (2016) The Ensembl Variant Effect Predictor. Genome Biol 17:122. https://doi.org/10.1186/s13059-016-0974-4
Acknowledgements
We thank Pauline Jones for her help with data and sample collection.
We thank NIHR BioResource volunteers for their participation, and gratefully acknowledge NIHR BioResource centres, NHS Trusts and staff for their contribution. We thank the National Institute for Health Research, NHS Blood and Transplant, and Health Data Research UK as part of the Digital Innovation Hub Programme. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. The research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. Funding sources are Kids Kidney Research; Nephrotic Syndrome Trust; Kidney Research UK; National Institute for Health Research (NIHR, grant number RG65966) and Guys and St Thomas’ Hospital Charity.
The UK Renal Rare Disease Registry (RaDaR) is funded by Kidney Research UK and British Kidney Patients’ Association. Drosophila work supported by Kidney Research UK and British Heart Foundation.
A.B. is funded by Kidney Research UK (Non-clinical Post-doctoral Fellowship).
This study was carried out on behalf of all the investigators in the UK National Study of Nephrotic Syndrome (NephroS).
Additional funding was from MRC Precision Medicine grant number MR/RO13942/1, and from Nephrotic Syndrome Trust (NeST).
Author information
Authors and Affiliations
Contributions
M.A.S, P.S.H, and A.B.K designed the study and A.B., K.B., S.M., P.D., C.N., E.C., H.J.M., A.B.K, S.H., C.B, M.D.S., K.S. K.M., C.P. R.M., S.M., N.F., M.A., H.S., M.W., G.I.W. recruited patients, gathered detailed clinical information for the study, carried out the experiments and/or analysed the data; A.B., K.B., P.S.H, and M.A.S. wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the Bristol Research Ethics Committee, REF 09/H0106/80.
Consent to participate
Consent for sequencing was obtained from all patients according to the Research Ethics.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bierzynska, A., Bull, K., Miellet, S. et al. Exploring the relevance of NUP93 variants in steroid-resistant nephrotic syndrome using next generation sequencing and a fly kidney model. Pediatr Nephrol 37, 2643–2656 (2022). https://doi.org/10.1007/s00467-022-05440-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-022-05440-5