
Abstract
Recent studies have demonstrated that levels of fibroblast growth factor 23 (FGF-23), a key regulator of phosphorus and vitamin D metabolism, rise dramatically as renal function declines and may play a key initiating role in disordered mineral and bone metabolism in patients with chronic kidney disease (CKD). The physiologic importance of FGF-23 in mineral metabolism was first identified in human genetic and acquired rachitic diseases and further characterized in animal models. FGF-23 and its regulators, including phosphate regulating endopeptidase homolog, dentin matrix 1 (DMP1), and matrix extracellular phosphoglycoprotein, are made primarily in bone, specifically in osteocytes. Dysregulation of these proteins results in osteomalacia, implicating the osteocyte in the regulation of skeletal mineralization. Studies in pediatric patients with CKD, the majority of whom have altered skeletal mineralization in early stages of CKD, have demonstrated that skeletal expression of both FGF-23 and its regulator, DMP1, are increased in early stages of CKD and that expression of these proteins is associated with alterations in skeletal mineralization. Thus, dysregulation of osteocytic proteins occur very early in the course of CKD and appear to be central to altered bone and mineral metabolism in this patient population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Abnormalities in mineral and bone metabolism occur early in the course of chronic kidney disease (CKD) and progress as renal function declines [1]. Traditionally, these abnormalities have been ascribed to changes in the parathyroid hormone (PTH) and vitamin D axis, which lead to subsequent alterations in calcium and phosphorus metabolism [1–3]. However, recent studies have revealed that circulating values of fibroblast growth factor 23 (FGF-23), a key regulator of phosphorus and vitamin D metabolism, rise dramatically as renal function declines and may play a key initiating role in the development of abnormal mineral metabolism in patients with CKD [4].
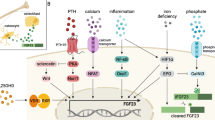
FGF-23 is made in osteocytes in mineralized bone [5], and studies of FGF-23 in human on genetic and acquired diseases and those using animal models have demonstrated that both under- and over-expression [6–8] of FGF-23 result in impairments in bone biology. Although the defective skeletal mineralization observed in patients with FGF-23 excess is likely a consequence of low phosphorus and vitamin D values, studies of FGF-23 deficiency in animal models and in cell culture suggest that FGF-23, and the proteins that regulate FGF-23, also have a direct effect on bone [9]. In these models, FGF-23 appears to directly regulate osteoblast differentiation [9], while a complete lack of the FGF-23 protein impairs skeletal mineralization, despite adequate (even excessive) circulating levels of phosphorus and vitamin D [6, 7]. In addition, recent studies suggest that alterations in skeletal FGF-23 expression also coincide with impairments in skeletal metabolism in the CKD population. Indeed, FGF-23 is up-regulated early in the course of CKD and is associated with skeletal mineralization indices in these individuals [10]. Although the mechanisms by which these effects on bone are mediated are unknown, they may involve a number of proteins that have been shown to regulate both FGF-23 levels and skeletal mineralization [11, 12].
FGF-23 and mineral metabolism: function and regulation
The physiologic importance of FGF-23 was first identified in human genetic and acquired rachitic diseases, such as autosomal dominant hypophosphatemic rickets (ADHR), tumor-induced osteomalacia (TIO), and X-linked hypophosphatemic rickets (XLH) [8, 13–15]. In these conditions, increased levels of the protein are accompanied by impaired tubular phosphate reabsorption, hypophosphatemia, low (or inappropriately normal) levels of 1,25 (OH)2vitamin D, and impaired skeletal mineralization (rickets or osteomalacia) [6, 7, 16–18]. Increased circulating levels of FGF-23 are necessary and sufficient to account for these abnormalities since the infusion of FGF-23 into rats with normal renal function results in renal phosphate wasting and direct suppression of 1α hydroxylase activity [18] while the inverse phenotype accompanies a complete lack of either functional FGF-23 or its co-receptor, Klotho [6, 7, 19–23]. More recently, FGF-23 has also been shown to regulate PTH metabolism based on observations that it can suppress PTH secretion both in vitro and in vivo [24, 25].
FGF-23 expression is regulated by vitamin D, phosphate and, potentially, PTH. In both animals and humans, the administration of 1,25(OH)2vitamin D increases circulating FGF-23 levels [26], apparently due to a direct action of vitamin D on FGF-23 via a vitamin D response element located upstream of the FGF-23 promoter [27]. Sustained increases in dietary phosphorus are also associated with increasing FGF-23 levels and declining 1,25(OH)2vitamin D levels [28, 29], while dietary phosphorus restriction reverses these trends [28, 29]. PTH levels may also stimulate FGF-23 expression [30]; findings in primary hyperparathyroidism [30], McCune–Albright syndrome [31], and Jansen’s disease [32] suggest that osteocytic stimulation by PTH directly increases skeletal FGF-23 release. The mechanism by which phosphate and PTH mediate changes in FGF-23 expression remain unknown and may be either direct effects on FGF-23 gene expression itself or mediated through other potential regulators of FGF-23.
Regulation of FGF-23: effects on bone metabolism and interaction with other skeletal proteins
Although the effects of FGF-23 on mineral metabolism obscure the potential direct effects of the protein on bone biology, a growing compendium of data from animals as well as from genetic and acquired human diseases of FGF-23 deficiency and excess have yielded many insights into the role that both FGF-23 and the factors that regulate FGF-23 play in bone biology. While FGF-23 is expressed in a variety of tissues, the majority of circulating FGF-23 is derived from osteocytes (in high levels) and osteoblasts (in lower levels) [33]. Although Klotho, the obligate co-receptor for the actions of FGF-23 on mineral metabolism, has not been described in skeletal tissue, a number of studies suggest that FGF-23 has a direct effect on bone.
FGF-23 appears to directly inhibit osteoblast maturation and matrix mineralization particularly during embryonic skeletal development [9]. Consistent with an effect of FGF-23 on osteoblast proliferation, FGF-23 expression is much lower in the embryonic skeleton than it is in adult animals [5] and, indeed, disruption of the Wnt signaling pathway—a pathway responsible for osteoblast proliferation and bone matrix mineralization—has been noted in mice with excess skeletal FGF-23 expression [34]. In mature animals, a complete lack of FGF-23 also results in focal alterations in skeletal mineralization, despite adequate (even excessive) serum phosphate, calcium, and vitamin D levels [6, 7], suggesting a direct role of the protein on maintaining skeletal mineralization at later stages of development.
Several factors have been described which are produced in bone and regulate skeletal FGF-23 expression and which may themselves contribute to the skeletal mineralization process. The genetic condition of XLH (a condition with a phenotype very similar to that of ADHR) and its mouse homolog, the Hyp mouse, are associated with increased FGF-23 levels as a result of defects in the phosphate-regulating endopeptidase homolog (PHEX). PHEX is a cell-surface endopeptidase predominantly located in osteoblasts and osteocytes. Although the exact actions of PHEX in vivo have not yet been completely defined, inactivation of PHEX leads to increased FGF-23 expression by an indirect mechanism. Potential mediators of these increased FGF-23 levels include increased skeletal expression of fibroblast growth factor 1, which has been shown to directly stimulate the FGF-23 promoter [34], and decreased expression of UDP-N-acetyl-alpha-D-galactosamine polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3), an enzyme essential for the glycosylation (and hence, stabilization) of the FGF-23 protein [34].
Whether from a direct effect of increased skeletal FGF-23 expression or due to some other factor modulated by the loss of PHEX activity, bone from Hyp mice displays an intrinsic mineralization defect that is not corrected by normalization of circulating calcium and phosphate concentrations; indeed, the selective ablation of PHEX in osteoblasts and osteocytes is sufficient to generate a phenotype of osteomalacia in mice [35], while the transplantation of Hyp mouse bone into wild-type mice does not reverse the phenotype of the explanted bone [36]. This intrinsic mineralization defect may be due to excessive proteolytic activity in the absence of PHEX; Rowe et al. [37] have demonstrated that the mineralization defect in the Hyp mouse can be reversed with CA074 and pepstatin—inhibitors of proteolytic activity—without correcting systemic hypophosphatemia. Factors which regulate local pH, such as carbonic anhydrase 12 (Car12), carbonic anhydrase 3 (Car3), and sodium-dependent citrate transporter (Slc13a5) expression, are also dysregulated in Hyp osteoblasts [34], suggesting that altered local bicarbonate and/or citrate concentrations may also impair mineralization by depriving the osteocyte of citrate necessary for energy metabolism. In addition, intrinsic mineralization inhibitors, including matrix gla protein (MGP) and thrombospondin (Thbs) 4, are increased in Hyp mouse osteocytes and may also contribute to altered skeletal mineralization [34].
Skeletal mineralization in various forms of hypophosphatemic rickets may also be regulated through interactions with members of the short integrin binding-ligand, N-linked Glycoprotein (SIBLING) family. It has been proposed that PHEX binds to members of the (SIBLING) family, proteins which regulate both FGF-23 [11] and the process of skeletal mineralization [38]. Indeed, PHEX regulates at least two SIBLING proteins, matrix extracellular phospho-glycoprotein (MEPE) [11] and dentin matrix protein 1 (DMP1), thereby preventing their proteolytic cleavage and the release their active C-terminal peptide [11, 12]. Consistent with these findings, MEPE is increased in XLH patients and in Hyp mice [11, 39], while PHEX inhibits the cleavage of the acidic, serine- and aspartic acid-rich motif (ASARM) peptide, an active peptide which inhibits mineralization, from full-length MEPE [11]. However, as the deletion of MEPE fails to correct the Hyp phenotype [39], other factors are likely to be involved.
The role of DMP1 in the regulation of FGF-23 and skeletal mineralization may be of even greater importance. In contrast to MEPE, DMP1, or rather the 2 active (N- and C-terminal) fragments of DMP1 generated from its cleavage by such proteinases as bone morphogenic protein 1 (BMP1) [40], promotes mineral formation [41]. In both humans and animals, DMP1 dysfunction results in increased skeletal and circulating FGF-23 values as well as a diffuse skeletal mineralization defect [33, 42] and disrupted osteocyte structure [33]. Furthermore, the DMP1/FGF-23 double knockout is phenotypically similar to the FGF-23 knockout [43], suggesting that DMP1 regulates FGF-23 and is located upstream of the FGF-23 molecule.
The effects of FGF-23 on mineral and bone metabolism in CKD
FGF-23 levels rise progressively as renal function declines [4, 44]. Several potential mechanisms for these increasing values have been proposed, including (1) an increased production by bone in response to a decreased capacity for renal phosphate excretion and/or (2) decreased renal clearance of FGF-23. The variety of assays available for detecting FGF-23 complicates this issue; currently, the “intact” molecule may be detected by two assays produced by two different manufacturers: Kainos and Immutopics [45]. A “C-terminal” assay is also available (Immutopics, Los Angeles, CA) that has the potential to measure potentially inactive C-terminal fragments of the molecule [45]. However, in a number of studies, values of FGF-23 by these different assays are well correlated [10, 46], and recent data suggest that the vast majority of circulating FGF-23 in dialysis patients is in the full-length, intact, active form of the molecule [47]. Thus, although these three assays are calibrated differently, all three likely measure full-length, active FGF-23 in circulation.
Regardless of the cause, increased FGF-23 values are found in early stages of CKD—before any abnormalities in serum calcium, phosphorus, or PTH are apparent [4, 48, 49]. Since normal serum phosphate levels are typically maintained until late in the course of CKD [4], increasing concentrations of FGF-23 appear to represent a compensatory response to maintain normal serum phosphate levels in the face of declining nephron mass. As FGF-23 values are independently associated with decreasing kidney function and low 1,25(OH)2vitamin D levels [4], the decline in calcitriol levels associated with increasing FGF-23 levels is thought to represent the initial event in the development of secondary hyperparathyroidism.
As in patients with primary excesses in FGF-23 [8], defective skeletal mineralization is also common in patients with all stages of chronic kidney disease, in whom increased circulating levels of FGF-23 occur in the presence of normal or elevated serum phosphorus values [6, 7]. However, the association between FGF-23 and bone in this population differs greatly from that in the general population. The results of a cross-sectional analysis of 49 pediatric dialysis patients with secondary hyperparathyroidism suggest that high circulating levels of FGF-23 in pediatric dialysis patients are associated with improved indices of skeletal mineralization [10]. Although these results appear to contrast with findings in patients with normal kidney function, they are similar to the mineralization defects found in rodents with a complete lack of FGF-23, despite adequate circulating mineral content [6, 7].
Confirming this association, a study of FGF-23, DMP1, and MEPE expression in bone tissue of 32 pediatric and young adult patients with CKD demonstrated that both FGF-23 and DMP1 expression were up-regulated in trabecular bone in early (stage 2) CKD, while MEPE expression remained unchanged from normal controls. In patients with all stages of CKD, the amount of bone FGF-23 correlated directly with bone DMP1 expression and the expression of each was inversely related to osteoid accumulation. In contrast, MEPE expression was not related to skeletal mineralization, but it was inversely related to bone volume. Although the simultaneous increase in both DMP1 and FGF-23 expression appears to be contrary to previous data suggesting that DMP1 acts to suppress FGF-23 expression, other data have suggested that the over-expression of DMP1 does not suppress FGF-23 expression [50]. Moreover, DMP1 promoter activity increases in response to increasing phosphate concentrations [51]. Thus, it is possible that the simultaneous increase in bone DMP1 and FGF-23 expression reflects the increasing phosphate burden associated with progressive renal failure. Alternatively, increased DMP1 expression may reflect an alteration in protein function in the context of CKD. Although the mechanism by which this might occur is unknown, alterations in DMP1 protein phosphorylation or cleavage [41] could play a role. Recent data suggest that DMP1 undergoes post-translational cleavage, leaving less than 1% of the protein in the full-length form [52]. The cleavage products appear to have distinct biological functions; in vitro mineralization studies have demonstrated that while the carboxyl-terminal fragment promotes mineralization [41, 53], the full-length DMP1 molecule may inhibit hydroxyapatite formation [41]. Thus, alterations in protein cleavage could have significant ramifications for DMP1 function.
Summary
FGF-23 plays a central role in mineral and bone metabolism. This role was initially delineated by the study of genetic and acquired conditions of hypophosphatemic rickets, but the greatest clinical impact of the discovery of FGF-23 may be in the management of CKD patients. FGF-23 and its regulators are made in osteocytes in bone, and in patients with CKD, FGF-23 levels rise as renal function declines, likely due to the decreasing capacity of the damaged kidney to excrete dietary phosphorus loads. Rising FGF-23 levels contribute to the development of secondary hyperparathyroidism and may also be linked to alterations in skeletal mineralization in the CKD population. Thus, through the expression of various proteins crucial to mineral metabolism, osteocytes appear to be endocrine cells with a key role in the regulation of skeletal mineralization. Alterations in osteocyte metabolism occur in very early stages of CKD and likely mediate altered bone and mineral metabolism in patients with even very mild degrees of renal dysfunction.
References
Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, Andress DL (2007) Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 71:31–38
National Kidney Foundation (2003) K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 42:S1–S201
National Kidney Foundation (2005) K/DOQI clinical practice guidelines for bone metabolism and disease in children with chronic kidney disease. Am J Kidney Dis 46:S1–S121
Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, Jüppner H, Wolf M (2005) Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 16:2205–2215
Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, Kozai K, Tanne K, Aubin JE, Maeda N (2007) Mineralized tissue cells are a principal source of FGF23. Bone 40:1565–1573
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568
Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Jüppner H, Lanske B (2004) Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 23:421–432
ADHR consortium (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, Aubin JE, Maeda N (2008) Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res 23:939–948
Wesseling-Perry K, Pereira RC, Wang H, Elashoff RM, Sahney S, Gales B, Jüppner H, Salusky IB (2009) Relationship between plasma FGF-23 concentration and bone mineralization in children with renal failure on peritoneal dialysis. J Clin Endocrinol Metab 94:511–517
Guo R, Rowe PS, Liu S, Simpson LG, Xiao ZS, Quarles LD (2002) Inhibition of MEPE cleavage by Phex. Biochem Biophys Res Commun 297:38–45
Martin A, David V, Laurence JS, Schwarz PM, Lafer EM, Hedge AM, Rowe PS (2008) Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 149:1757–1772
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 98:6500–6505
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Jüppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348:1656–1663
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S (2002) Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87:4957–4960
White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren O, Meitinger T, Strom TM, Jüppner H, Econs MJ (2001) The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab 86:497–500
De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY, Kumar R, Levine MA, Schiavi SC (2002) Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res 17:1102–1110
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429–435
Kato K, Jeanneau C, Tarp MA, et-Pages A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H (2006) Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem 281:18370–18377
Araya K, Fukumoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T (2005) A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab 90:5523–5527
Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B (2005) An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 14:385–390
Larsson T, Yu X, Davis SI, Draman MS, Mooney SD, Cullen MJ, White KE (2005) A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab 90:2424–2427
Bai X, Dinghong Q, Miao D, Goltzman D, Karaplis AC (2009) Klotho ablation converts the biochemical and skeletal alterations in FGF23 (R176Q) transgenic mice to a Klotho-deficient phenotype. Am J Physiol Endocrinol Metab 296:E79–E88
Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerstrom G, Jonsson KB, Westin G, Larsson TE (2007) Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 195:125–131
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro O, Mohammadi M, Sirkis R, Naveh-Many T, Silver J (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117:4003–4008
Yu X, Sabbagh Y, Davis SI, Demay MB, White KE (2005) Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone 36:971–977
Barthel TK, Mathern DR, Whitfield GK, Haussler CA, Hopper HA, Hsieh JC, Slater SA, Hsieh G, Kaczmarska M, Jurutka PW, Kolek OI, Ghishan FK, Haussler MR (2007) 1, 25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J Steroid Biochem Mol Biol 103:381–388
Antoniucci DM, Yamashita T, Portale AA (2006) Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab 91:3144–3149
Burnett SA, Gunawardene SC, Bringhurst FR, Jüppner H, Lee H, Finkelstein JS (2006) Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 21:1187–1196
Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y (2007) Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol 18:2683–2688
Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron RP (2003) FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 112:683–692
Brown WW, Jüppner H, Langman CB, Price H, Farrow EG, White KE, McCormick KL (2009) Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen's metaphyseal chondrodysplasia. J Clin Endocrinol Metab 94:17–20
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z, Quarles LD (2009) Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol 23:1505–1518
Yuan B, Takaiwa M, Clemens TL, Feng JQ, Kumar R, Rowe PS, Xie Y, Drezner MK (2008) Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest 118:722–734
Liu S, Tang W, Zhou J, Vierthaler L, Quarles LD (2007) Distinct roles for intrinsic osteocyte abnormalities and systemic factors in regulation of FGF23 and bone mineralization in Hyp mice. Am J Physiol Endocrinol Metab 293:E1636–E1644
Rowe PS, Matsumoto N, Jo OD, Shih RN, Oconnor J, Roudier MP, Bain S, Liu S, Harrison J, Yanagawa N (2006) Correction of the mineralization defect in hyp mice treated with protease inhibitors CA074 and pepstatin. Bone 39:773–786
Addison WN, Nakano Y, Loisel T, Crine P, McKee MD (2008) MEPE-ASARM peptides control extracellular matrix mineralization by binding to hydroxyapatite: an inhibition regulated by PHEX cleavage of ASARM. J Bone Miner Res 23:1638–1649
Liu S, Brown TA, Zhou J, Xiao ZS, Awad H, Guilak F, Quarles LD (2005) Role of matrix extracellular phosphoglycoprotein in the pathogenesis of X-linked hypophosphatemia. J Am Soc Nephrol 16:1645–1653
Maciejewska I, Cowan C, Svoboda K, Butler WT, D'Souza R, Qin C (2009) The NH2-terminal and COOH-terminal fragments of dentin matrix protein 1 (DMP1) localize differently in the compartments of dentin and growth plate of bone. J Histochem Cytochem 57:155–166
Tartaix PH, Doulaverakis M, George A, Fisher LW, Butler WT, Qin C, Salih E, Tan M, Fujimoto Y, Spevak L, Boskey AL (2004) In vitro effects of dentin matrix protein-1 on hydroxyapatite formation provide insights into in vivo functions. J Biol Chem 279:18115–18120
Lorenz-Depiereux B, Bastepe M, et-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM (2006) DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 38:1248–1250
Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD (2008) Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol Metab 295:E254–E261
Larsson T, Nisbeth U, Ljunggren O, Jüppner H, Jonsson KB (2003) Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 64:2272–2279
Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM, Shulman D, Kassem M, Rackoff P, Zimering M, Dalkin A, Drobny E, Colussi G, Shaker JL, Hoogendoorn EH, Hui SL, Econs MJ (2006) Sensitivity of fibroblast growth factor 23 measurements in tumor-induced osteomalacia. J Clin Endocrinol Metab 91:2055–2061
Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M (2008) Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 359:584–592
Shimada T, Urakawa I, Isakova T, Yamazaki Y, Epstein M, Wesseling-Perry K, Wolf M, Salusky IB, Jüppner H (2009) Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab. doi:10.1210/jc.2009-1603
Shigematsu T, Kazama JJ, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, Fukagawa M (2004) Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis 44:250–256
Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Kronenberg F, Kuen E, Konig P, Kraatz G, Mann JF, Muller GA, Kohler H, Riegler P (2007) Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 18:2600–2608
Lu Y, Ye L, Yu S, Zhang S, Xie Y, McKee MD, Li YC, Kong J, Eick JD, Dallas SL, Feng JQ (2007) Rescue of odontogenesis in Dmp1-deficient mice by targeted re-expression of DMP1 reveals roles for DMP1 in early odontogenesis and dentin apposition in vivo. Dev Biol 303:191–201
Lu Y, Liu S, Xie Y, Yu S, Quarles L, Bonewald LF, Feng JQ (2007) Use of the transgenic approach to determine the role of DMP1 in phosphate regulation. J Musculoskelet Neuronal Interact 7:309
Huang B, Maciejewska I, Sun Y, Peng T, Qin D, Lu Y, Bonewald L, Butler WT, Feng J, Qin C (2008) Identification of full-length dentin matrix protein 1 in dentin and bone. Calcif Tissue Int 82:401–410
He G, Gajjeraman S, Schultz D, Cookson D, Qin C, Butler WT, Hao J, George A (2005) Spatially and temporally controlled biomineralization is facilitated by interaction between self-assembled dentin matrix protein 1 and calcium phosphate nuclei in solution. Biochemistry 44:16140–16148
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wesseling-Perry, K. FGF-23 in bone biology. Pediatr Nephrol 25, 603–608 (2010). https://doi.org/10.1007/s00467-009-1384-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-009-1384-6