
Abstract
Isolated caprine early-staged follicles were submitted to osmotic tolerance tests in the presence of sucrose, ethylene glycol (EG), or NaCl solutions and were exposed to and cryopreserved (by slow or rapid cooling) in MEM alone or MEM supplemented with sucrose, EG (1.0 or 4.0 M), or both. When follicles were exposed to 1.5 M NaCl, only 2% of the follicles were viable, whereas 87% of the follicles were viable after exposure to 4.0 M EG. Regarding exposure time, the highest percentage of viable follicles was obtained when follicles were exposed for 10 min to 1.0 M EG + 0.5 M sucrose; exposure for 60 s to 4.0 M EG + 0.5 M sucrose also maintained high percentage viability in follicles. Slow cooling in the presence of 1.0 M EG + 0.5 M sucrose (75%) or rapid cooling in the presence of 4.0 M EG + 0.5 M sucrose (71%) resulted in a significantly higher proportion of viable follicles than all other treatments (P < 0.05). A 24-h culture of frozen-thawed follicles was used to assess survival; only slow-frozen follicles showed viability rates similar to control follicles (64% vs. 69% respectively; P > 0.05). Interestingly, the percentage of viable rapid-cooled follicles (59%) was similar to that obtained after in vitro culture of conventional slow-cooled follicles but was significantly lower than that in controls. Thus, in addition to determining improved procedures for the exposure of follicles to EG and sucrose before and after freezing of caprine early-staged follicles, we report the development of rapid- and slow-cooling protocols.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The major aim of our study has been to test several combinations of factors associated with a cryopreservation procedure by using slow- and rapid-cooling methods for early-staged caprine follicles. The factors that indirectly influence the cryo-survival of cells in general fall into two categories: the internal properties inherent to the specific individual cells of the tissues of interest, and the external factors involved in all essential steps of the cryopreservation procedure (Aubard et al. 1994). Internal properties include the geometry of individual cells, the permeability of individual cells and of the entire follicle to water and cryoprotectants at various temperatures, and the developmental stage of the follicle. External factors that should be considered include the way that the material is treated prior to cooling, the composition of solutions including the type and method of addition of the cryoprotectant, the cooling rate and the final super-cooling and transfer to liquid nitrogen, the thawing rate, and the rate and method of cryoprotectant removal/dilution.
Our group has previously developed two cryopreservation protocols (slow and rapid cooling) to store caprine ovarian fragments (Santos et al. 2006a, 2007a). Our rationale for the present study is to improve these protocols further in order to obtain better preservation of the gametes enclosed in early-staged follicles (primordial, primary, and secondary follicles), the major source of oocytes in the ovary (Gougeon 1993). Improved storage of such follicles might be useful for endangered breeds or could, at a later stage, be applied to rescue oocytes from human patients subjected to chemotherapy treatments for cancer. Furthermore, humans and small ruminants possess ovaries and follicles of similar size, morphology, and structure (Lucci et al. 2001).
Successful cryopreservation protocols developed for ovarian tissue may not be suitable for isolated cells or groups of cells, such as follicles. In contrast to larger parts of ovarian tissues, follicles lack the protection of a connective tissue barrier. This absence is likely to influence the intrinsic properties of the tissue, particularly with regard to an increased permeability to cryoprotectants and water. Damage caused by cryoprotectants is often associated with the shrinkage and swelling of the cell in response to highly concentrated solutions (Mazur et al. 2000). During exposure to high concentrations of cryoprotectants (equilibrium phase), cryoprotective agents move across cell membranes because of the concentration gradient, and this flow may impair cell viability (Agca et al. 2002). Several explanations for damage to cells resulting from cryoprotectant exposure include damage via hyper-osmotic changes (Agca et al. 2000), cryoprotectant removal (Santos et al. 2007a), or the composition of the cryoprotectant solutions (Stachecki et al. 1998). Cell damage can be caused by chemical and osmotic toxicity. Cryoprotectants are claimed to cause cell damage because they are used at concentrations that exceed physiological osmolality levels (280–300 mOsm/kg). However, previous studies have shown that high concentrations of electrolytes are correlated with cellular damage (Lovelock 1953), and cryoprotectants are non-ionic solutes. Thus, we wanted to compare cryoprotectant solutions with a solution easily dissociable into ions (NaCl).
Cryoprotectant removal and a return to isotonic conditions may cause cell damage (Santos et al. 2007a), as does adverse temperature exposure (De la Cueva et al. 1997). Exposure of early-staged follicles to cryoprotectant solutions is often performed at 20°C, and after freezing-thawing, cryoprotectant removal is performed to permit cells to return to isotonic conditions (Rodrigues et al. 2004a, b). Osmotic tolerance (i.e., the ability to survive exposure to high solute concentrations) has been suggested to be dependent on temperature, since less osmotic damage is observed at low temperatures such as 4°C (Steponkus 1984). Furthermore, sperm cells appear to maintain membrane integrity if they are not returned to isotonic conditions after exposure to cryoprotectants (Armitage and Mazur 1984). This might be attributable to the method of cryoprotectant removal (in a graduated series or by direct exposure of cells to a cryoprotectant-free medium). In addition, the viability of cryopreserved early-staged follicles may be compromised after thawing and subsequent culture; we have demonstrated that 24-h in vitro culture of frozen-thawed caprine ovarian follicles is an essential step in the evaluation of the cryopreservation process (Santos et al. 2006a).
The aim of this study has been to determine the influence of various external factors so as to improve cryopreservation of early-staged caprine follicles. The factors tested include iso- and anisotonic solutions (NaCl, sucrose and EG) at two different exposure temperatures (4°C and 20°C), and the effect on viability of intra-cellular (EG) and extra-cellular (sucrose) cryoprotectants. These variables have been compared by using both a conventional slow-freezing protocol and a rapid-freezing protocol.
Materials and methods
This study was composed of four different experiments. For each experiment, female reproductive organs from five adult (1–2 years old) mixed-breed goats were obtained at a local slaughterhouse. Ovarian pairs were transported at 30°C to the laboratory within 1 h and then washed once in 70% alcohol for 10 s and twice in phosphate-buffered saline (PBS). The cortex from each pair of ovaries was then removed and cut into fragments (1 mm3). The fragments were placed in PBS supplemented with 0.1% (v/v) penicillin/streptomycin (Gibco, Paisley, UK) at room temperature (RT), and then the follicles were isolated by using a mechanical procedure described by Lucci et al. (1999). Only early-staged follicles smaller than 100 µm in diameter were used and transferred to HEPES-buffered M199 + 1% bovine serum albumin (Sigma, St. Louis, Mo., USA). Every treatment, from each experiment, was repeated four times.
Slow cooling
Groups of ∼25 early-staged follicles were placed in an eye-dish plate containing 1 ml cryopreservation medium (see experiment 3) and then loaded into 1 ml Cassou straws (IMV, L’Aigle, France). Straws were first rinsed by aspirating freezing medium and expelling it immediately, followed by aspiration of freezing medium containing the early-staged follicles. Subsequently, straws were sealed with humidified polyvinylalcohol powder, placed into the controlled-rate programmable freezer (Planer Kryo 10 Series II, Cryotech Benelux, Schagen, The Netherlands), and incubated for 10 min at 20°C. Samples were cooled as described by Santos et al. (2006a), after which the straws were immersed into liquid nitrogen (−196°C) and stored for 1 week. When required, the straws were thawed in air for 1 min at RT and then immersed in a water bath at 37°C until the cryopreservation medium had completely melted, after which the cryoprotectant was removed in three steps by incubating the samples in MEM with 0.3, 0.15, and 0.0 M sucrose for 5 min each.
Rapid cooling
Groups of ∼25 early-staged follicles were placed in 1 ml straws containing the cryopreservation medium, as described for slow cooling, and incubated at 20°C for 60 s (see experiment 3). Immediately after the incubation period, straws were immersed vertically into liquid nitrogen and stored in liquid nitrogen for 1 week. The samples were thawed as described for slow cooling. Based on results from experiment 2, cryoprotectant was removed by washing three times for 5 min each in sucrose-free MEM solution.
In vitro culture
In vitro culture was performed in 4-well culture dishes (Nunc, Roskilde, Denmark) with 25 follicles per well, in a 20-µl droplet of culture medium under mineral oil at 39°C, in a humidified chamber with 5% CO2 in air. The culture medium consisted of alpha MEM (Sigma) supplemented with SPIT (5 ng/ml selenium, 110 µg/ml pyruvate, 10 µg/ml insulin, and 5.5 µg/ml transferrin; Sigma), 100 µg/ml penicillin/streptomycin (Gibco), 10% fetal calf serum (Gibco), 0.01 IU/ml recombinant human follicle-stimulating hormone (hFSH; Organon, Oss, The Netherlands), and 10 ng/ml activin-A (R&D System Europe, Abingdon, UK).
Trypan blue staining
The viability of the follicles was evaluated by the addition of 5 µl 0.4% trypan blue (Sigma) to each 100 µl solution and incubation for 1 min (RT) as described by Jewgenow et al. (1998). Follicles were classified as non-viable when ≥10% of the cells were stained with trypan blue and viable when <10% of the cells were stained with trypan blue. As a positive control, follicles were treated with Triton X-100 for 3 min and stained with trypan blue (see Supplementary Fig. 1).
Ethidium/calcein staining
Follicular viability was also evaluated by incubating the follicles in a mixture of 4 µM calcein AM, 2 µM ethidium homodimer-1 (Molecular Probes Europe, Leiden, The Netherlands), and 10 µM Hoechst 33342 (Sigma), as described by Santos et al. (2006a). Oocytes and granulosa cells were classified as viable if the ooplasm was stained positively with calcein, and chromatin was unlabeled with ethidium. The percentage of viable granulosa cells was calculated in relation to the total number of Hoechst-positive nuclei (Supplementary Fig. 2). Viable follicles contained a viable oocyte surrounded by ≥90% viable granulosa.
Experiment 1: osmotic tolerance of early-staged follicles to NaCl, sucrose, and EG
Early-staged follicles were submitted to hypertonic conditions caused by intra- (1.0 or 4.0 M EG) and extra-cellular (0.5 M sucrose) cryoprotectant solutions or 1.5 M NaCl and to a hypotonic condition caused by 0.015 M NaCl. To show the effect of cryoprotectant removal, follicles exposed to EG and sucrose were immediately evaluated with trypan blue, or washed in MEM or in MEM supplemented with a series of gradually decreasing sucrose concentrations (0.3, 0.15, and 0.0 M), and then stained. As controls, follicles were evaluated immediately after isolation or exposed to a 0.9% (0.15 M) NaCl solution. Groups of 20 follicles were subjected to each osmotic treatment with solutions of different osmolalities (28, 282, and 2800 mOsm/kg of NaCl, 0.015, 0.15, and 1.5 M, respectively; 792 mOsm/kg sucrose, 0.5 M; 2726 and 5242 mOsm/kg EG, 1.0 and 4.0 M, respectively) and one isotonic solution (0.9% NaCl, 286 mOsm/kg). Exposure was performed in 100-µl drops of the solutions for 10 min at 4°C or 20°C (Supplementary Scheme 1). In the sucrose and EG treatments, follicles were analyzed either immediately or after being washed in MEM (Sigma) alone or in MEM supplemented with 0.3, 0.15, and 0.0 M sucrose.
Experiment 2: effect of dose and exposure length to cryoprotectants on early-staged follicle viability prior to freezing
Sixteen aliquots (each containing ∼25 isolated follicles) were incubated at 20°C for 1, 5, 10, or 20 min in 1 ml MEM with or without 0.5 M sucrose, 1.0 M EG, or both. Another 16 aliquots were incubated for 15, 30, 60, or 120 s in 1 ml MEM (Sigma) with or without 0.5 M sucrose, 4.0 M EG, or both (Supplementary Scheme 2). After incubation, the follicles from each group were washed in three steps (5 min each) to remove cryoprotectant, based on the best results from experiment 1. After cryoprotectant removal, follicles were analyzed by trypan blue staining.
Experiment 3: comparison between slow and rapid cooling of early-staged follicles
Follicles (20 per treatment) were submitted to slow cooling in MEM alone or to the addition of sucrose, EG (1.0 M), or both. Rapid cooling of follicles was performed in MEM alone or in MEM with added sucrose, EG (4.0 M), or both (based on optimal conditions observed in experiment 2; see Supplementary Scheme 3). All cryopreservation procedures were performed in the absence of serum, and the duration of exposure of follicles to cryoprotectants before freezing was based on results from experiment 2 (10 min for slow and 60 s for rapid cooling). After 1 week, samples were thawed, cryoprotectant was removed, and follicles were evaluated by using ethidium homodimer and calcein as viability markers.
Experiment 4: in vitro culture of early-staged follicles
To compare both freezing procedures, as determined according to experiment 3, fresh (immediately isolated from fresh ovaries), slow-, or rapid-cryopreserved early-staged follicles (25 follicles per treatment) were evaluated immediately or after in vitro culture for 24 h. Follicular analysis was performed by using ethidium homodimer and calcein-AM as viability markers (Supplementary Scheme 4).
Statistical analysis
The mean percentage of viable follicles and the viability of oocytes and granulosa cells were compared by a one-way analysis of variance (ANOVA) and Tukey’s test by using Statview for Windows. Differences were considered significant when P≤0.05.
Results
Experiment 1: osmotic tolerance of early-staged follicles to NaCl, sucrose, and EG
At both 4°C and 20°C, follicles were spherical under isotonic conditions (0.15 M NaCl), whereas they shrunk after exposure to high osmolality NaCl (1.5 M NaCl), or to 0.5 M sucrose, or to 1.0 M, and 4.0 M EG. In contrast to cells incubated in NaCl (1.5 M) or sucrose (0.5 M), cells maintained in EG regained their volume to some extent during exposure (Fig. 1). After exposure to 1.5 M NaCl at 4°C and 20°C, only 2% of follicles were unstained, indicating that shrinkage resulted in the loss of membrane integrity (Fig. 2). In contrast, hypertonic solutions of sucrose or EG did not cause the same deleterious effect as 1.5 M NaCl solution. Exposure to 0.5 M sucrose (4°C) or 1.0 M EG (4°C and 20°C) resulted in a higher percentage of viable follicles when cryoprotectant was gradually removed by step-wise washing in MEM supplemented with decreasing sucrose concentrations than when follicles were washed in MEM alone (Fig. 2). In contrast, follicles exposed to 4.0 M EG at both temperatures were damaged less when cryoprotectant was removed in MEM only. No significant difference was observed in the percentage of follicular viability between exposure temperatures, except in 4.0 M EG in which viability was higher at 20°C (Fig. 2).

Changes in follicular volume imposed during hyper-osmotic treatments. Images indicate changes in volume of primary follicles compared with the original volume (dotted lines at 0 sec) during exposure to 1.5 M NaCl (a–f), 4.0 M ethylene glycol (EG, g–l), and 0.5 M sucrose (Sucr., m–r). Only the follicle exposed to 4.0 M EG retained its original volume (g–l). Bars 40 µm

Viability based on trypan blue staining of isolated caprine early-staged follicles exposed to NaCl (experiment 1; n = 150 follicles per repetition) or cryoprotectant (0.5 M sucrose, or 1.0 or 4.0 M EG; experiment 1; n = 300 follicles per repetition) solutions. *Differs significantly from control (P < 0.05). Values with different letters (a–c) differ significantly among the NaCl solution concentrations (0.09%, 0.9%, and 9%) within the same temperature (P < 0.05). Values with different lower-case letters (a, b) differ significantly among the effects of exposure only or washing treatments after exposure to the cryoprotectant solutions (MEM alone or MEM supplemented with a series of gradually decreasing sucrose concentrations; P < 0.05). Values with different upper-case letters (A, B) differ significantly between 4°C and 20°C with the same cryoprotectant solution (P < 0.05)
Experiment 2: effect of dose and exposure length to cryoprotectants on early-staged follicle viability prior to freezing
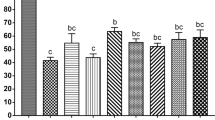
Based on experiment 1, a second experiment was performed to determine the optimal duration of follicle exposure to cryoprotectant. Thirty-four groups of follicles were analyzed after exposure to MEM or cryoprotectant. Among non-exposed control follicles, 93% were viable (trypan-blue-negative). Exposure of follicles to MEM + 0.5 M sucrose did not affect their viability; this was also observed when follicles were exposed to 1.0 M EG or 1.0 M EG + 0.5 M sucrose for up to 10 min. Longer exposure (20 min) to 1.0 M EG with or without 0.5 M sucrose resulted in a significant decrease in the percentage of viable follicles (Fig. 3a). The majority of non-exposed follicles were viable (89%). Exposure to MEM or MEM + 0.5 M sucrose for 120 s did not affect follicular viability. Viability was also similar to controls when follicles were exposed to MEM supplemented with 4.0 M EG or 4.0 M EG + 0.5 M sucrose for up to 60 s. Compared with 1.0 M EG, the window of optimal cryoprotectant exposure was dramatically reduced when 4.0 M EG was used (Fig. 3a,b).

Viability based on trypan blue staining of isolated caprine early-staged follicles exposed to MEM, sucrose (0.5 M), or EG at 1.0 M (a) or to MEM, sucrose (0.5 M), or EG at 4.0 M (b; experiment 2a, b; n = 200 follicles per repetition). +Differs significantly from control and from other treatments (P < 0.05). *Differs significantly from control (P < 0.05). Values with different lower-case letters (a, b) differ significantly among treatments within the same exposure time (P < 0.05). Values with different upper-case letters (A, B) differ significantly between exposure times in the same cryoprotectant (P < 0.05)
Experiment 3: comparison between slow and rapid cooling of early-staged follicles
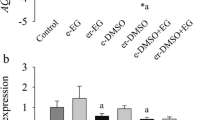
Using the results from experiment 2, we assessed the effect of slow- versus rapid-cooling methods of cryopreservation on the viability of early-staged follicles. Viability was evaluated by using ethidium homodimer and calcein-AM. All freezing treatments significantly reduced the percentages of viable early-staged follicles when compared with controls, except for slow cooling in a mixture of EG and sucrose (Fig. 4a,b). The highest percentage of viable follicles was obtained when early-staged follicles were slowly cooled in MEM containing 0.5 M sucrose and 1.0 M EG, whereas the most rapidly cooled follicles appeared viable when early-staged follicles were frozen in MEM containing 4.0 M EG (with or without sucrose). Both these most optimal cryopreservation methods resulted in similar follicular survival rates (Fig. 4a,b).

Viability based on ethidium-homodimer and calcein-AM labeling of isolated caprine early-staged follicles after conventional (slow) cooling (a) and rapid cooling (b; experiment 3a, b; n = 150 follicles per repetition). Viable follicles had a viable oocyte surrounded by ≥90% viable granulosa cells. *Differs significantly from control (P < 0.05). Values with different letters (a–d) differ significantly among the treatments (P < 0.05)
Experiment 4: in vitro culture of early-staged follicles
Based on experiment 3, isolated follicles were cultured in vitro for 24 h before (fresh) or after freeze-thawing with the best slow- and rapid-cooling conditions. In vitro culture decreased the viability of fresh follicles. After culture, the percentage of viable oocytes and follicles among slowly cooled follicles did not significantly differ from those of fresh follicles. When compared with fresh follicles, the percentages of viable oocytes and follicles among rapidly cooled follicles decreased significantly during culture, whereas no changes were detected for slowly cooled follicles (Fig. 5).

Percentage of fresh and frozen-thawed viable oocytes and early-staged follicles before and after in vitro culture for 24 h (experiment 4; n = 120 follicles per repetition). Viable follicles had a viable oocyte surrounded by ≥90% viable granulosa cells. *Differs significantly from control (P < 0.05). Values with different lower-case letters (a, b) differ significantly between cryopreservation procedures before or after culture within the same tissue (P < 0.05). Values with different upper-case letters (A, B) differ significantly before and after in vitro culture for 24 h within the same freezing procedure (P < 0.05)
Discussion
We describe methods to improve the preservation of caprine early-staged follicles. In this regard, we have tested the effect of different osmotic pressures on those follicles, the effect of slow and rapid freezing in MEM supplemented with cryoprotectants such as sucrose, EG, or a mixture of both, or the effect of instant versus step-wise removal of these cryoprotectants.
Exposure to hypotonic and hypertonic solutions of NaCl reduced the viability of early-staged follicles, both at 4°C and 20°C. We found that more damage was caused by high sodium concentrations than by high solute concentrations per se or by exposure temperature. Mouse zygotes exposed to high osmolar solutions (1,200–2,000 mOsm/kg) of PBS supplemented with choline chloride remain intact after cooling and re-warming, whereas PBS supplemented with sodium chloride was lethal to the embryos subjected to the same regime (Toner et al. 1993). Therefore, although rarely stated, high solute concentrations are not necessarily damaging (Stachecki et al. 1998). In addition to simple diffusion, eukaryotic cells have mechanisms by which they counteract changes in osmolality, such as the Na+/K+ pump, which plays a role in the maintenance of intracellular osmotic conditions (Darnell et al. 1986).
Although EG is known for its low toxicity when used in cryopreservation procedures of caprine and bovine ovarian tissue (Santos et al. 2006a, 2007a; Celestino et al. 2007), other cryoprotectants such as dimethyl sulfoxide preserves the ultrastructure of sheep early-staged follicles better than EG (Santos et al. 2006b). This illustrates the fundamental requirement for an understanding of the chemical pathways of cryoprotectant-cell interaction to explain specific differences. Our findings show that, apart from exposure to EG or sucrose, loss of follicular viability is also caused by the procedure of cryoprotectant removal, which has been shown to be damaging to other cell systems (Karlsson and Toner 1996). The optimal way to reacquire isotonic equilibrium after exposure is obtained by washing samples in solutions containing decreasing concentrations of sucrose (except when incubating follicles in 4.0 M EG). The high concentration of cryoprotectant (making it essential to avoid toxic effects by removing the cryoprotectant as quickly as possible by direct washes in MEM alone) or prolonged exposure to anisotonic condition might cause viability loss. Thus, EG might be more toxic under osmotic stress. In an additional experiment, we have studied the effect on early-staged follicles of the cryoprotectant exposure, since the loss of viability in follicles can occur prior to freezing (Rodrigues et al. 2004a, b). Early-staged follicles cannot be exposed to EG for extended period of time (>10 min for 1.0 M EG, and >60 s for 4.0 M EG). Changes in membrane stability increase the permeability of membranes for water and cations, leading to a higher susceptibility to damage. To control these changes, exposure to cryoprotectant solutions must be improved by using a carbohydrate such as sucrose (an extra-cellular cryoprotectant) that is usually used as an osmotic buffer to prevent excessive volume changes.
A simple medium (serum-free single intra-cellular EG supplemented with an extra-cellular cryoprotectant, viz., sucrose) was used instead of a mixture of two or several intra-cellular cryoprotective compounds. This approach has previously been successful in slow-cooling studies with caprine early-staged follicles enclosed in ovarian tissue (Santos et al. 2006a). Human embryos have been cryopreserved in EG-sugar based solutions (Ohta et al. 1996). Although EG is not the most optimal glass former (Fahy et al. 2004), it is the most indicated cryoprotectant for the cryopreservation of goat and human ovarian follicles. Furthermore, sucrose has been often successfully used in cryopreservation procedures (Chung et al. 2000).
Slow- and rapid-cooled follicles in a mixture of EG and sucrose exhibit similar viability. However, after culturing, only slow cooling results in viability similar to those of fresh follicles, whereas rapid cooling leads to a marginal but significant decrease in viability. The maintenance of morphology and viability is a major problem in the in vitro culture of early-staged follicles from large mammals (Santos et al. 2007b). Although the method studied for rapidly cooling the follicles is not as successful as the slow-cooling method, continued fundamental studies should attain better methods that might make a difference with regard to the preservation of genetic material from endangered breed animals. These animals may be encountered dead and far from laboratories equipped with freezing machines, thus illustrating the importance of a simple freezing method with no need of sophisticated equipment.
Our findings on osmotic tolerance, cryoprotectant removal, cryoprotectant exposure, slow vs. rapid cooling, and post-thaw culture of isolated goat early-staged follicles show that the most suitable protocol, among those that we have tested for post-thaw follicular survival, consists of exposure to MEM supplemented with 1.0 M EG and 0.5 M sucrose for 10 min, followed by slow-cooling and subsequent cryoprotectant removal with sucrose in a series of decreasing concentrations (0.3, 0.15, and 0.0 M). We should mention that, in our experiments, frozen-thawed follicles have been analyzed in vitro after a post-thaw culture for 24 h. Although this approach has helped to improve the freeze-thawing protocol, the in vivo survival and development of follicles frozen-thawed with the described methods demands further study.
References
Agca Y, Liu J, Rutledge JJ, Critser ES, Critser JK (2000) Effect of osmotic stress on the developmental competence of germinal vesicle and metaphase II stage bovine cumulus oocyte complexes and its relevance to cryopreservation. Mol Reprod Dev 55:2112–2119
Agca Y, Gilmore J, Byers M, Woods EJ, Liu J, Critser JK (2002) Osmotic characteristics of mouse spermatozoa in the presence of extenders and sugars. Biol Reprod 67:1493–1501
Armitage WJ, Mazur P (1984) Osmotic tolerance of human granulocytes. Am J Physiol 247:373–381
Aubard Y, Teissier MP, Baudet JH (1994) Cryopreservation of the ovary and ovarian tissue. Rev Fr Gynecol Obstet 89:192–197
Celestino JJ, Santos RR, Lopes CA, Martins FS, Matos MH, Melo MA, Báo SN, Rodrigues AP, Silva JR, Figueiredo JR (2007) Preservation of bovine preantral follicle viability and ultra-structure after cooling and freezing of ovarian tissue. Anim Reprod Sci (in press)
Chung HM, Hong SW, Lim JM, Lee SH, Cha WT, Ko JJ, Han SY, Choi DH, Cha KY (2000) In vitro blastocyst formation of human oocytes obtained from unstimulated and stimulated cycles after vitrification at various maturational stages. Fertil Steril 73:545–551
Darnell J, Lodis H, Baltimore D (1986) Active transport of ions and ATP hydrolysis. In: Darnell J, Lodis H, Baltinore D (eds) Molecular cell biology. Scientific American Books, New York, pp 625–628
De la Cueva FI, Pujol MR, Rigau T, Bonet S, Miro J, Briz M, Rodriguez-Gill JE (1997) Resistance to osmotic stress of horse spermatozoa: the role of ionic pumps and their relationship to cryopreservation success. Theriogenology 48:947–968
Fahy GM, Wowk B, Wu J, Paynter S (2004) Improved vitrification solutions based on the predictability of vitrification solution toxicity. Cryobiology 48:22–35
Gougeon A (1993) Dynamics of human follicular growth: a morphologic perspective. In: Adashi EY, Leung PCK (eds) The ovary. Raven, New York, pp 21–39
Jewgenow K, Penfold LM, Meyer HH, Wildt DE (1998) Viability of small preantral ovarian follicles from domestic cats after cryoprotectant exposure and cryopreservation. J Reprod Fertil 112:39–47
Karlsson JOM, Toner M (1996) Long-term storage of tissues by cryopreservation: critical issues. Biomaterials 17:243–256
Lovelock JE (1953) The mechanism of the protective action of glycerol against haemolysis by freezing and thawing. Biochem Biophys Acta 11:28–36
Lucci CM, Amorim CA, Bao SN, Figueiredo JR, Rodrigues APR, Silva JRV, Gonçalves PBD (1999) Effect of the interval of serial sections of ovarian tissue in the tissue chopper on the number of isolated caprine preantral follicles. Anim Reprod Sci 56:39–49
Lucci CM, Silva JRV, Carvalho FCA, Figueiredo JR, Bao SN (2001) Light microscopical and ultrastructural characterization of goat preantral follicles. Small Rum Res 41:61–69
Mazur P, Katkov II, Katkova N, Critser JK (2000) The enhancement of the ability of mouse sperm to survive freezing and thawing by the use of high concentrations of glycerol and the presence of an Escherichia coli membrane preparation (oxyrase) to lower the oxygen concentration. Cryobiology 40:187–209
Ohta N, Nohara M, Kojimahara T (1996) Ultrarapid freezing of human embryos by vitrification method. Jpn J Fertil Steril 41:276–279
Rodrigues AP, Amorim CA, Costa SH, Matos MHT, Santos RR, Lucci CM, Bao SN, Ohashi OM, Figueiredo JR (2004a) Cryopreservation of caprine ovarian tissue using glycerol and ethylene glycol. Theriogenology 61:1009–1024
Rodrigues AP, Amorim CA, Costa SH, Matos MHT, Santos RR, Lucci CM, Bao SN, Ohashi OM, Figueiredo JR (2004b) Cryopreservation of caprine ovarian tissue using dimethyl sulfoxide and propanediol. Anim Reprod Sci 84:211–227
Santos RR, Tharasanit T, Figueiredo JR, Van Haeften T, Van den Hurk R (2006a) Preservation of caprine preantral follicles viability after cryopreservation in sucrose and ethylene glycol. Cell Tissue Res 325:523–531
Santos RR, Rodrigues APR, Costa SHF, Silva JRV, Matos MHT, Lucci CM, Bao SN, Van den Hurk R, Figueiredo JR (2006b) Histological and ultrastructural analysis of cryopreserved sheep preantral follicles. Anim Reprod Sci 91:249–263
Santos RR, Tharasanit T, Van Haeften T, Figueiredo JR, Silva JRV, Van den Hurk R (2007a) Vitrification of goat preantral follicles enclosed in ovarian tissue by using conventional and solid-surface vitrification methods. Cell Tissue Res 327:167–176
Santos RR, Van den Hurk R, Rodrigues APR, Costa SHF, Martins FS, Matos MHT, Celestino JJH, Figueiredo JR (2007b) Effect of cryopreservation on viability, activation and growth of in situ and isolated ovine early-staged follicles. Anim Reprod Sci 99:53–64
Stachecki JJ, Cohen J, Willadsen SM (1998) Cryopreservation of unfertilized mouse oocytes: the effect of replacing sodium with choline in the freezing medium. Cryobiology 37:346–354
Steponkus PL (1984) Role of the plasma membrane in freezing injury and cold acclimation. Annu Rev Plant Physiol 35:543–584
Toner M, Cravalho EG, Stachecki J, Fitzgerald T, Tompkins RG, Yarmush ML, Armant DR (1993) Nonequilibrium freezing of one-cell mouse embryos: membrane integrity and developmental potential. Biophys J 64:1908–1921
Acknowledgements
The authors thank Arend Rijneveld and Frans van Kooi for logistical help, Dr. Damien Paris for the English review, and two anonymous reviewers for their helpful suggestions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the image is a link to a high resolution version
Supplementary Figure 1
Viability of goat early-staged follicles based on trypan blue staining (blue staining non-viable cells). a Positive control, Triton X-100 treated secondary follicle. b Viable secondary follicle. c Non-viable secondary follicle. Bars 50 μm (JPG 240 kb)
Supplementary Figure 2
Viability of goat early-staged follicles based on staining with Hoechst (blue staining all nuclei) and ethidium homodimer-1 (red staining nuclei from dead cells). a, b Representative image of a viable primordial follicle. c, d Representative image of two non-viable primordial follicles. Follicle 1 contains a viable oocyte, whereas fewer than 90% of granulosa cells are viable. Follicle 2 contains three oocytes of which one has deteriorated, and fewer than 90% of granulosa cells are viable (arrowheads oocytes). Note that polyovular healthy early-staged (primordial, primary, and secondary) follicles are commonly observed in goats (Lucci et al. 1999). Bars 30 μm (JPG 301 kb)
Supplementary Scheme 1
Experiment 1: osmotic tolerance of early-staged follicles to NaCl, sucrose, and ethylene glycol (EG) (DOC 29 kb)
Supplementary Scheme 2
Experiment 2: dose and duration dependency of early-staged follicles exposed to cryoprotectants (suc sucrose) prior to freezing (DOC 33 kb)
Supplementary Scheme 3
Experiment 3: comparison between slow and rapid cooling of early-staged follicles (suc sucrose) (DOC 33 kb)
Supplementary Scheme 4
Experiment 4: in vitro culture of early-staged follicles (suc sucrose) (DOC 34 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Santos, R.R., van Haeften, T., Roelen, B.A.J. et al. Osmotic tolerance and freezability of isolated caprine early-staged follicles. Cell Tissue Res 333, 323–331 (2008). https://doi.org/10.1007/s00441-008-0613-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-008-0613-9