
Abstract
Most patients with congenital anomalies of the kidney and urinary tract (CAKUT) remain genetically unexplained. In search of novel genes associated with CAKUT in humans, we applied whole-exome sequencing in a patient with kidney, anorectal, spinal, and brain anomalies, and identified a rare heterozygous missense variant in the DACT1 (dishevelled binding antagonist of beta catenin 1) gene encoding a cytoplasmic WNT signaling mediator. Our patient’s features overlapped Townes–Brocks syndrome 2 (TBS2) previously described in a family carrying a DACT1 nonsense variant as well as those of Dact1-deficient mice. Therefore, we assessed the role of DACT1 in CAKUT pathogenesis. Taken together, very rare (minor allele frequency ≤ 0.0005) non-silent DACT1 variants were detected in eight of 209 (3.8%) CAKUT families, significantly more frequently than in controls (1.7%). All seven different DACT1 missense variants, predominantly likely pathogenic and exclusively maternally inherited, were located in the interaction region with DVL2 (dishevelled segment polarity protein 2), and biochemical characterization revealed reduced binding of mutant DACT1 to DVL2. Patients carrying DACT1 variants presented with kidney agenesis, duplex or (multi)cystic (hypo)dysplastic kidneys with hydronephrosis and TBS2 features. During murine development, Dact1 was expressed in organs affected by anomalies in patients with DACT1 variants, including the kidney, anal canal, vertebrae, and brain. In a branching morphogenesis assay, tubule formation was impaired in CRISPR/Cas9-induced Dact1−/− murine inner medullary collecting duct cells. In summary, we provide evidence that heterozygous hypomorphic DACT1 variants cause CAKUT and other features of TBS2, including anomalies of the skeleton, brain, distal digestive and genital tract.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) comprise various malformations resulting from defects in the morphogenesis of the kidneys and/or the urinary tract. Kidney anomalies observed within the CAKUT spectrum range from severe manifestations, such as kidney agenesis or multicystic dysplastic kidney (MCDK), to milder phenotypes, including kidney hypoplasia or fused/duplex kidney (Schedl 2007). Taken together, all CAKUT phenotypes have a prevalence of 3–9/1000 live births (Pohl et al. 2002; Queisser-Luft et al. 2002; Stoll et al. 2014), and account for around 40% of cases with end-stage kidney disease in children and adolescents (Harambat et al. 2012). CAKUT occur sporadically in around 85% of patients. In familial cases, inheritance is often dominant. Over 500 syndromes are associated with CAKUT (Limwongse 2009), and around one-third of patients present with extrarenal features (Stoll et al. 2014).
To date, around 60 genes are known to cause isolated or mild syndromic CAKUT in humans if mutated (Kosfeld et al. 2018; van der Ven et al. 2018b). Chromosomal aberrations (Stoll et al. 2014) including microdeletions/-duplications (Weber et al. 2011; Sanna-Cherchi et al. 2018) may also be causative. Despite increasing knowledge about the genetic basis of human CAKUT, the majority of patients remain genetically unexplained (van der Ven et al. 2018a, b). Although identifying new CAKUT-causing genes remains challenging due to high genetic heterogeneity, variable expressivity and incomplete penetrance (van der Ven et al. 2018b), gene discovery has been accelerated by the advent and large-scale use of next generation sequencing (NGS) technologies. Dominant genes associated with human CAKUT using NGS include DSTYK (Sanna-Cherchi et al. 2013), TBX18 (Vivante et al. 2015), TBC1D1 (Kosfeld et al. 2016), PBX1 (Heidet et al. 2017), GREB1L (Brophy et al. 2017; De Tomasi et al. 2017), LIFR (Kosfeld et al. 2017; Christians et al. 2020), TBX6 (Verbitsky et al. 2019; Yang et al. 2020), GDF6 (Martens et al. 2020), and ZMYM2 (Connaughton et al. 2020); recessive genes include ITGA8 (Humbert et al. 2014) and ROBO1 (Münch et al. 2022).
NGS techniques have been especially successful in identifying the underlying genetic cause of syndromic CAKUT patients (van der Ven et al. 2018a). In this study, whole-exome sequencing (WES) in a patient presenting with unilateral kidney agenesis and contralateral duplex kidney as well as malformations of the spine, distal digestive tract, and central nervous system yielded a very rare heterozygous variant in the DACT1 (dapper, dishevelled binding antagonist of beta catenin 1) gene. DACT1 is a known murine CAKUT gene (Suriben et al. 2009; Wen et al. 2010) encoding a cytoplasmic protein acting in WNT signaling (Cheyette et al. 2002; Zhang et al. 2006). A DACT1 nonsense variant was described in a family with features overlapping Townes–Brocks syndrome 1 (TBS1, OMIM # 107480) (Webb et al. 2017) referred to as TBS2 (OMIM # 617466). By studying the frequency, clinical impact, and functional consequences of DACT1 variants in a cohort of CAKUT patients, investigating Dact1 expression during murine development, and analyzing the consequences of Dact1 deficiency in an in vitro model of tubulogenesis, we provide further evidence that Dact1 deficiency and very rare DACT1 variants may cause kidney and specific extrarenal anomalies in mice and humans.
Patients and methods
Patients
The study was approved by the Ethics Boards of Hannover Medical School, Hannover, Germany; Tübingen University Hospital, Tübingen, Germany; Oslo University Hospital, Oslo, Norway; Skopje University Hospital, Skopje, North Macedonia. Each family provided informed consent for participation in the study. Of the 209 CAKUT patients analyzed, 130 were males, 79 were females, and their mean age was 11.5 years (range 2–37 years). All 209 patients had kidney anomalies, 56 were additionally affected by vesicoureteral reflux, and 33 patients had undergone kidney transplantation due to end-stage kidney disease. The spectrum of kidney anomalies with or without urinary tract malformations of the analyzed patients is listed in Supplementary Table 1. Case reports of patients carrying DACT1 variants are provided in the supplementary material.
Whole-exome and targeted DACT1 sequencing
WES was performed on leukocyte DNA of 38 CAKUT patients and 137 individuals not affected by CAKUT (serving as in-house controls for WES data analysis of index patient V005-II.04, Supplementary Table 2) using the SureSelectXT Human All Exon V4 target enrichment kit (Agilent, Santa Clara, CA, USA) on a HiSeq 2000 (Illumina, San Diego, CA, USA) sequencer or the SureSelectXT Human All Exon V5 + UTRs target enrichment kit (Agilent) on a HiSeq 2500 (Illumina) sequencer. All samples were sequenced to a mean coverage of 50x. Sequencing data were aligned to the human reference genome (hg19/GRCh37) using the CLC Genomics Workbench (version 5.0.2; Qiagen, Hilden, Germany). WES data were annotated and prioritized using Ingenuity Variant Analysis (Qiagen) and our in-house NGS data analysis workflow. Supplementary Table 2 summarizes the candidate gene-based strategy used to analyze WES data of index patient V005-II.04. Using conventional chain termination protocols and a 3130XL Genetic Analyzer (Life Technologies, Carlsbad, CA, USA), mutational analysis of all coding exons and adjacent intronic regions of NM_016651.5(DACT1) was done to verify variants identified by WES analysis, to determine familial segregation, and to screen for DACT1 variants in 171 further CAKUT patients. Supplementary Table 3 summarizes the sequences of oligonucleotides used. The minor allele frequencies (MAF) of genetic variants were retrieved from the Genome Aggregation Database (gnomAD controls v2.1.1, total population, https://gnomad.broadinstitute.org/). Variant pathogenicity was predicted using CADD (https://cadd.gs.washington.edu/snv; Kircher et al. 2014; Rentzsch et al. 2019), MutationTaster (http://www.mutationtaster.org/), SIFT (https://sift.bii.a-star.edu.sg/), PROVEAN (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and classified using the ACMG guidelines (Richards et al. 2015).
Animals
All applicable international, national and/or institutional guidelines for the care and use of animals were followed. All experiments were approved by the Ethics Board of the Lower Saxony State Office for Consumer Protection and Food Safety. Murine embryos were derived from matings of Ztm:NMRI wildtype mice. For timed pregnancies, vaginal plugs were checked in the morning after mating, and noon was defined as embryonic day (E) 0.5. Embryos or urogenital systems were dissected in phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde (PFA) in PBS followed by subsequent dehydration in methanol. Fixed embryos or urogenital systems were stored in 100% methanol at −20 °C prior to RNA in situ hybridization analysis.
RNA in situ hybridization on sections of murine embryos or kidneys
To determine the expression pattern of Dact1 during murine embryonic development, non-radioactive RNA in situ hybridization analysis was carried out following a standard protocol (Moorman et al. 2001). In brief, PFA-fixed embryos or urogenital systems of wildtype mice were paraffin-embedded, and sectioned to 10 µm thickness. Sections were deparaffinized in Carl Roth ROTI Histol (#10379029; Thermo Fisher Scientific, Waltham, MA, USA), sequentially rehydrated in ethanol/H2O, washed in PBS, and treated with 10 µg/ml proteinase K (#7528; Carl Roth, Karlsruhe, Germany) in 0.1 M Tris, pH 8.0 at 37 °C for 8 min. After washing with 0.2% glycerin/PBS and PBS, and post-fixation with 4% PFA / 0.2% glutaraldehyde at room temperature (RT) for 20 min each, sections were hybridized with a digoxygenin-labeled riboprobe (DIG RNA Labeling Mix, #11277073910; Sigma-Aldrich, St. Louis, MO, USA) directed against mouse Dact1 mRNA (514 bp, NM_001190466, position 872-1385) in hybridization buffer at 70 °C overnight. Sections were washed twice in 50% formamide / 50% 2xSSC (pH 7.0) at 65 °C for 20 min. Probes were detected using Anti-Digoxigenin-AP, Fab fragments (2 h at RT) and BM-Purple AP substrate (#11093274910 and #11442074001; Sigma-Aldrich). For each developmental stage, at least 3 specimens were analyzed. Stained sections were documented on a Leica DM5000 microscope using a Leica DFC300 FX digital camera (Leica Microsystems, Wetzlar, Germany).
Cloning of expression constructs and site-directed mutagenesis
To generate a DACT1 expression construct, the full-length DACT1 open reading frame was amplified from human cDNA and sub-cloned into the pcDNA3.1-Myc vector (Thermo Fisher Scientific) using customized oligonucleotides (Supplementary Table 3) and the In-Fusion HD Cloning Kit (Takara Bio, Kusatsu, Japan). The variants were inserted into the DACT1 expression construct using customized oligonucleotides (Supplementary Table 3) and the Phusion Site-Directed Mutagenesis Kit (Thermo Fisher Scientific).
Cell culture and transient transfection
Human embryonic kidney 293T (HEK293T) cells were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM; Merck, Darmstadt, Germany) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (all Thermo Fisher Scientific). For murine inner medullary collecting duct 3 (mIMCD3) cells, DMEM/Ham's F-12 (1:1) medium (Merck) was used. Cell cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2. For transient transfection of HEK293T cells, Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific) was used following standard protocols.
Immunoprecipitation
To analyze binding of wildtype and mutant Myc-DACT1 to Flag-DVL2 (dishevelled segment polarity protein 2) by immunoprecipitation (IP), HEK293T cells (1.0 × 107) were seeded in Petri dishes and transiently co-transfected with pcDNA3.1-Myc-DACT1 (wildtype or mutant) and pCMV5-Flag(3x)-DVL2 (#24802; Addgene, Watertown, MA, USA). At 24 h post transfection, cells were lysed in IP buffer (50 mM Tris-HCl, pH 8.0, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1% Triton X-100) supplemented with protease and phosphatase inhibitors (Roche Diagnostics, Mannheim, Germany). After adding 1 µg of anti-Myc antibody (#sc-40; Santa Cruz Biotechnology, Dallas, TX, USA), lysates were rotated overnight at 4 °C. Protein G Sepharose beads (GE, Boston, MA, USA) were equilibrated in IP buffer and incubated with the lysates for 4 h at 4 °C. After washing 5 × with IP lysis buffer, proteins eluted from the beads using Laemmli buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerin, 2% sodium dodecyl sulfate (SDS), 5% 2-mercaptoethanol, 1 mM ethylenediaminetetraacetic acid, 0.01% bromophenol blue) were detected by Western blot analysis.
Western blot analysis
After SDS-polyacrylamide gel electrophoresis and semi-dry electro-blotting, nitrocellulose membranes (GE) were treated with 5% fat-free milk powder dissolved in PBS with 0.05% Tween 20 (PBST) as blocking agent. The primary antibodies mouse anti-Myc (#sc-40; Santa Cruz Biotechnology) or mouse anti-Flag (#8146; Cell Signaling Technology, Danvers, MA, USA) were diluted at 1:1,000 in 5% (w/v) bovine serum albumin in PBST and used for immunodetection. After incubation overnight at 4 °C, membranes were washed with PBST, exposed to the secondary horseradish peroxidase-conjugated anti-mouse antibody (#sc-2962; Santa Cruz Biotechnology; dilution 1:3,000) in 5% fat-free milk powder dissolved in PBST for 90 min at RT, washed again with PBST, and developed using the SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific). Signals were acquired using the Fusion FX7 gel documentation system (Vilber, Collégien, France). Densitometric quantification of protein bands was performed using ImageJ software (Schneider et al. 2012).
Qualitative DACT1 mRNA expression analysis
To determine whether mIMCD3 cells express Dact1, RNA was isolated from mIMCD3, HEK293T (positive control), and HeLa (negative control) cells (estimation of RNA expression levels based on www.proteinatlas.org) using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from RNA samples using the Superscript IV First-Strand Synthesis System (Thermo Fisher Scientific). Exon-spanning oligonucleotides specific for murine Dact1 cDNA (NM_021532.4, c.390-518) or human DACT1 cDNA (NM_016651.6, c.487-709) were used for PCR amplification (Supplementary Table 3), respectively. Sequencing of the generated amplicons was done using conventional chain termination protocols, as described above.
CRISPR/Cas9 genomic engineering
To generate a Dact1 knockout cell model, mIMCD3 cells and a protocol for CRISPR/Cas9-mediated RNA-guided genome editing (Ran et al. 2013) were used. In brief, a single guide RNA (sgRNA) targeting the first exon of Dact1 (targeted sequence 5’-GCG TAC CCG CGA GCG CCA GG-3’) was designed using the CRISPOR web-based tool (http://crispor.tefor.net), and sense and antisense oligonucleotides (Supplementary Table 3) were synthesized (Eurofins Genomics, Ebersberg, Germany). The dimerized oligonucleotides were inserted into a BpiI-digested pSpCas9(BB)-2A-GFP plasmid (#48138; Addgene), containing a sgRNA scaffold and expression cassettes for Cas9 and GFP. By transient transfection, the resulting construct was introduced into mIMCD3 cells. GFP-positive cells were isolated 24 h after transfection at the Cell Sorting Core Facility of Hannover Medical School using a MoFlo XDP cell sorter (Beckman-Coulter, Brea, MA, USA). To identify the genotype of selected cell clones, their DNA was extracted using the innuPREP DNA Mini Kit (Analytik Jena, Jena, Germany), and PCR products of Dact1 exon 1 were analyzed by direct sequencing (oligonucleotides listed in Supplementary Table 3). For allele-specific sequence analysis, the PCR product was cloned into a pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA) using oligonucleotides listed in Supplementary Table 3, and the plasmid DNA of at least 10 Escherichia coli transformants were analyzed by direct sequencing. In the three mIMCD3 cell clones selected for further analysis harboring either Dact1 wildtype (Dact1+/+; clone 2) or a biallelic knockout (Dact1−/−; clones 11 and 12), all 13 coding off-target sites were analyzed by direct sequencing (oligonucleotides given in Supplementary Table 3) to ascertain absence of mutation.
Tubulomorphogenesis assay
To investigate the consequences of a knockout of Dact1 on branching morphogenesis, a tubulomorphogenesis assay was performed using mIMCD3 cells, as previously described (De Tomasi et al. 2017). In brief, mIMCD3 cells were cultured in a three-dimensional (3-D) gel of collagen type I from rat tail (Corning, Corning, NY, USA) in 12-well plates. For each experiment, each cell clone was plated in duplicate. A thin collagen layer without cells was applied to the well, followed by a collagen layer containing 100,000 cells/ml. After the gel had solidified, 500 µl of DMEM/Ham's F-12 (1:1) medium (Merck) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 µg/ml streptomycin (all purchased from Thermo Fisher Scientific) were added to the well. After seven days of cultivation, cells were documented using an inverted microscope (DM IL LED Fluo, Leica Microsystems) equipped with an EC3 camera (Leica Microsystems). For better visualization, 3-D cultures were fixed in 4% PFA in PBS and stained with Alexa Fluor 488 Phalloidin (#A12379; Invitrogen; dilution 1:200 in PBST). For quantification, images of 3-D cultures were blinded, and at least 60 cellular structures were classified as tubular or spherical for each cell clone in each experiment (n = 3).
Statistical analysis
Statistical significance was calculated using Student’s t test or Fisher’s exact test (two-tailed), whereby p values of ≤ 0.05 were considered significant, and p values of ≤ 0.01 highly significant.
Results
Very rare heterozygous DACT1 variants predicted to be deleterious were identified in eight of 209 (3.8%) families with kidney anomalies
Under the assumption that NGS techniques are especially successful in identifying the genetic cause in syndromic CAKUT patients, we performed WES on leukocyte DNA of a four-year-old male index patient, V005-II.04, the child of non-consanguineous Kurdish parents (Fig. 1), born with a caudal regression syndrome including kidney, anorectal, and spinal anomalies and brain malformations (Fig. 2, Table 1). Kidney ultrasound, magnetic resonance imaging, and isotope nephrography were notable for left-sided kidney agenesis and a right-sided malrotated duplex kidney with hydronephrosis and primary obstructive megaureter (Fig. 2a–d). Additionally, a caudal regression syndrome with missing coccyx, sacral dysplasia, syringohydromyelia, an intraspinal dermoid cyst, anorectal agenesis with rectourethral fistula, and neurogenic bladder were diagnosed (Fig. 2e, f). In the central nervous system, agenesis of the septum pellucidum, triventricular hydrocephalus internus due to aqueductal stenosis, and agenesis of the cerebellar vermis were noted (Fig. 2g, h). Further details are provided in the supplementary material. WES data were analyzed using a candidate gene-based strategy and our in-house NGS data analysis pipeline. By prioritizing good-quality, non-silent, and very rare (MAF ≤ 0.0005) variants not present in in-house control individuals and known or presumed to cause isolated or syndromic CAKUT in humans or mice (Supplementary Table 2), we identified a very rare (MAF = 0.000333 according to gnomAD controls) missense variant in the DACT1 gene, NM_016651.5(DACT1):c.1100C > A p.(Thr367Lys). This variant is predicted to be deleterious by SIFT, PROVEAN, and PolyPhen-2, and has a CADD score of close to 20 (19.40) predicting that it is among the top 1.15% most deleterious variants in the human genome. The variant was confirmed to be heterozygous and shown to be inherited from the patient’s unaffected mother by targeted sequencing (Fig. 1). No other heterozygous or biallelic variants of interest, especially none in known or presumed CAKUT-associated genes in humans or mice, were identified in the patient. Considering that a family with a DACT1 nonsense variant (Webb et al. 2017) and Dact1 knockout mice (Suriben et al. 2009; Wen et al. 2010) had similar phenotypes involving the urogenital system, distal digestive tract, and spine, the DACT1 variant was considered to be causative and to explain the phenotype of our patient.

Identification of very rare heterozygous DACT1 missense variants in eight of 209 CAKUT families (3.8%). Electropherograms of DACT1 variants (affected base positions are indicated by arrows), their segregation, and the localization of affected amino acid residues within a representation of the DACT1 protein are shown. All seven different variants are located within the region of DVL2 interaction (*) according to Zhang et al. (2006). In pedigrees, squares denote males, circles females, and colored symbols affected individuals with phenotypes as indicated. Six of eight (75%) index CAKUT patients (indicated in pedigrees by an arrow) that received reverse phenotyping presented with extrarenal features including distal digestive tract anomalies, skeletal, genital and/or neurological anomalies. Black question marks denote family members with no clinical information available or without kidney ultrasound. Black circle with white question mark denotes individual with putative left-sided non-obstructive duplex kidney. Individual V005-II.04 was analyzed by whole-exome sequencing. Validation of the variant, identification of variants in further patients, and segregation analysis were performed by direct sequencing. + , represents DACT1 wildtype sequence; n.d., individual with non-available DNA

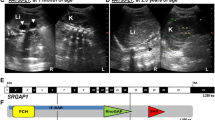
Phenotype spectrum of patients with DACT1 variants. a–h The index patient, V005-II.04, presented with numerous anomalies, including left-sided kidney agenesis (yellow arrow) and right-sided malrotated duplex kidney (ki) (white arrow) on magnetic resonance imaging (MRI) scan (a). By kidney ultrasound (US), the duplex kidney (marked by a dashed line, b) shows a dilated kidney pelvis (rp, c, d). By MRI scan, a caudal regression syndrome with missing coccyx and sacral dysplasia (yellow arrow, e), syringohydromyelia (sh) at T11-T12 (white arrow, f), and an intraspinal dermoid cyst (dc) at L2-L3 (yellow arrow, f) were diagnosed, along with a triventricular hydrocephalus internus (ventricles I-III, yellow arrows, g, h) due to an aqueductal stenosis, and an agenesis of the septum pellucidum (white arrow, h) in the central nervous system. i, j Patient T004-II.03 presented with bilateral kidney dysplasia on kidney US (right-sided kidney with hydronephrosis shown, i), and dilated kidney pelvises, megaureters (mu), and megacystis (mc) upon micturating cysto-urethrogram (j). k By kidney US of patient H452-II.01, a right-sided kidney hypoplasia and a left-sided normal kidney were diagnosed. l Patient H402-II.01 presented with right-sided kidney hypoplasia with a cyst (cy) and normal left-sided kidney on kidney US. m, n Kidney US of patient B036-II.01 showed a left-sided dilated residual ureter (ur) ending in a ureterocele (uc) located in the bladder (bl). o–q Patient N032-II.01 presented with a lumbarization of S1 (black arrow, o) on spinal X-ray (T12 is marked), no radiotracer uptake equivalent to a missing functional kidney on the left due to a multicystic dysplastic kidney (residues of which are seen as a cystic structure next to the bladder on US, p) and normal uptake in the right kidney on dimercaptosuccinic acid (DMSA) kidney scan (q)
To determine the frequency of DACT1 variants in a cohort of CAKUT patients, 208 additional families with kidney malformations (Supplementary Table 1) were subjected to WES or targeted DACT1 sequencing. We identified very rare (MAF ≤ 0.0005 according to gnomAD controls) missense variants in seven further families (Fig. 1). The variant carried by the index patient, c.1100C > A p.(Thr367Lys), was detected in a second patient (Fig. 1, Table 1). Two variants, c.2005C > G p.(Pro669Ala) and c.2468T > G p.(Leu823Arg), are not listed in the gnomAD database and have a CADD score ≥ 20, indicating that they are considered to be among the top 1% most deleterious variants in the human genome (Table 1). All variants except c.1703G > A p.(Arg568Lys) were deleterious according to at least two of five prediction tools (i.e., CADD, MutationTaster, SIFT, PROVEAN, PolyPhen2). Four variants were classified as likely pathogenic according to the ACMG guidelines, while three variants were of uncertain significance (Table 1). Each variant except c.1890G > T p.(Lys630Asn) was significantly more frequent in our cohort of CAKUT families compared to gnomAD controls (Table 1). Taken together, very rare (MAF ≤ 0.0005) non-silent DACT1 variants were found in eight of 209 (3.8%) families with kidney anomalies compared to 1,006 of 60,146 (1.7%) individuals from the gnomAD control cohort. This difference is statistically significant (p = 0.03, two-tailed Fisher’s exact test). Moreover, in three families we observed co-segregation of DACT1 variants with CAKUT or extrarenal phenotypes, i.e., megacystis in T004-II.03 and T004-II.04, kidney anomalies in H402-I.02 and H402-II.01, and skeletal anomalies in N032-I.02 and N032-II.01 (not all family members were available for genetic testing or phenotypic evaluation; Fig. 1, Supplementary Table 4). The DACT1 variants were either maternally inherited (6/8 families) or inheritance could not be determined (2/8 families; Fig. 1).
DACT1 variants convey a characteristic phenotype consisting of kidney plus anorectal, genital, skeletal or neurological anomalies in three quarters of CAKUT patients carrying DACT1 variants
In a reverse phenotyping effort, clinical or radiological reevaluation was performed of the CAKUT index patients from the eight families carrying very rare non-silent DACT1 variants. All eight patients presented with kidney phenotypes, i.e., unilateral kidney agenesis and contralateral malrotated duplex kidney with hydronephrosis (1/8 patients), unilateral multicystic dysplastic kidney (MCDK) (3/8), bilateral kidney dysplasia with or without cysts (2/8), or unilateral kidney hypoplasia with or without cysts (2/8) (Table 1, Fig. 2). Four patients were additionally diagnosed with anomalies of the urinary tract, i.e., megaureter (1/8), megaureter, vesicoureteral reflux (VUR) and megacystis (1/8), blind ending ureter (1/8), or VUR (1/8) (Table 1, Fig. 2). Notably, six of the eight (75%) CAKUT patients harboring DACT1 variants also presented with extrarenal anomalies similar to those described in a family with a heterozygous DACT1 loss-of-function variant (Webb et al. 2017) and in Dact1-deficient mice (Suriben et al. 2009; Wen et al. 2010). These include anomalies of the distal digestive tract, e.g., anorectal agenesis with recto-urethral fistula (1/8), genital features, e.g., anomalies of the uterus and ovary (1/8), skeletal features, e.g., spinal and craniofacial anomalies (3/8), and/or neurological features, e.g., malformations of the central nervous system, intellectual disability, autism (4/8) (Table 1, Fig. 2). Further details are provided in the supplementary material. Conversely, gastrointestinal, genital, skeletal or neurological anomalies were only detected in 64 of the 198 (32%) CAKUT patients without very rare non-silent DACT1 variants of whom information was available. Therefore, CAKUT patients with versus without very rare non-silent DACT1 variants were significantly more likely to present with extrarenal features in the digestive or genital tracts, skeleton or central nervous system (6/8, 75% versus 64/198, 32%; p = 0.02, two-tailed Fisher’s exact test).
Dact1 expression during early murine development was detected in organs showing defects in Dact1-deficient mice and patients carrying DACT1 variants
Dact1-deficient mice (Suriben et al. 2009; Wen et al. 2010) and patients carrying DACT1 variants (Webb et al. 2017) (Fig. 2, Table 1) show developmental defects that belong to the caudal regression syndrome including caudal vertebrae agenesis, anal atresia, kidney malformations, aberrantly ending ureters, bladder agenesis, and genital anomalies. In some patients carrying DACT1 variants, malformations of the brain and/or intellectual disability were also observed (Webb et al. 2017) (Fig. 2, Table 1). Detailed analysis of Dact1 expression in affected structures, especially in the kidney, during development has not been performed previously. Therefore, we analyzed the spatial and temporal expression of Dact1 in wildtype mouse embryos using RNA in situ hybridization on whole-embryo sections at E11.5, E12.5, and E14.5 (Fig. 3a), and on kidney sections at E16.5 and E18.5 (Fig. 3b). We detected Dact1 expression in organs of the caudal region including vertebrae, anal canal, kidney, bladder, genital tubercle as well as in the brain, spinal ganglia, inner ear, lung, and ribs (Fig. 3a). Expression in the upper urinary tract was confined to the mesenchyme of the ureter, and to the capsular, cortical, and medullary stroma of the kidney. Expression at these sites strongly decreased after E14.5 (Fig. 3a, b). Dact1 expression during early murine development is, therefore, observed in organs showing anomalies in Dact1-deficient mice and patients carrying DACT1 variants.

Dact1 expression pattern in murine embryonic development by non-radioactive RNA in situ hybridization. a On sections of murine embryos at E11.5, E12.5 and E14.5, Dact1 mRNA was detected in a variety of organs, including the kidney, bladder, anal canal, genital tubercle, lung, inner ear, brain, spinal ganglia, vertebrae, and ribs. Expression in the upper urinary tract was found in the mesenchyme of the ureter at E11.5, but not in the metanephric mesenchyme. At E12.5 and E14.5, Dact1 was additionally expressed in the capsular, cortical, and medullary stroma of the kidney. Similarly, in bladder, urethra, and anal canal Dact1 expression was confined to mesenchymal cells. b On kidney sections at E16.5 and E18.5, Dact1 expression, which starts to be reduced at E16.5 and is strongly diminished at E18.5, was detected in the mesenchyme of the ureter, the kidney capsule, and the stroma of medulla and cortex (higher magnification images). a, adrenal gland; ac, anal canal; b, brain; bl, bladder; gt, genital tubercle; ie, inner ear; k, kidney; l, lung; r, ribs; s, spinal ganglia; u, ureter; v, vertebrae. For each embryonic stage, at least 3 specimens were analyzed. Scale bars are as indicated
Dact1 knockout impairs tubule formation in a cellular model of branching morphogenesis
To characterize the impact of DACT1 on a process of major relevance for kidney development, we analyzed its role in a 3-D tubulomorphogenesis assay using mIMCD3 cells, a cellular model of branching morphogenesis (Chen et al. 2004; Mai et al. 2005; De Tomasi et al. 2017). Murine IMCD3 cells undergo tubulogenesis in a 3-D collagen gel (Chen et al. 2004), a process disrupted after knockout of relevant genes, such as GREB1L (De Tomasi et al. 2017). Here, in mIMCD3 cells, shown to express Dact1 by RT-PCR (Supplementary Fig. 1), a Dact1 knockout cell line was generated using CRISPR/Cas9 technology. For subsequent analysis, we selected a wildtype clone without mutational event at the sgRNA on-target site (clone 2, Dact1+/+), and two knockout clones with different biallelic Dact1 frameshift variants (clones 11 and 12, Dact1−/−) predicted to result in truncated non-functional proteins (Supplementary Fig. 2). As depicted in Fig. 4a and b, over 70% of unmodified mIMCD3 cells and clone 2 (Dact1+/+) control cells formed elongated tubular structures after seven days in a 3-D collagen gel, whereas both Dact1−/− cell clones failed to form tubules and grew as spherical structures. These data provide evidence that DACT1 is involved in tubulogenesis in vitro.

Characterization of Dact1-deficient mIMCD3 cells and DACT1 mutant proteins. a, b To analyze the relevance of DACT1 for tubulogenesis, a process of major relevance for kidney development, Dact1−/− mIMCD3 cells (clones 11 and 12) and control cells (clone 2, Dact1+/+), generated by CRISPR/Cas9 technology (Supplementary Fig. 2), were cultured in a 3-D collagen I matrix for seven days and stained with Alexa Fluor 488 phalloidin (scale bar represents 200 µm) (a). Quantification of the tubulomorphogenesis assay showed that more than 70% of mIMCD3 and clone 2 (Dact1+/+) cells developed tubular structures, whereas both Dact1−/− cell lines (clone 11 and clone 12) displayed nearly no tubuli. At least 60 structures were counted and rated as tubular or spherical for each cell line in each experiment (mean ± SD of three independent experiments) (b). c, d To explore the pathogenicity of the identified DACT1 variants, interaction of mutant DACT1 with DVL2 was analyzed by co-immunoprecipitation (IP) (c). The ratio of Flag-DVL2 to wildtype or mutant Myc-DACT1 in the immunoprecipitates was significantly decreased for all mutants compared to wildtype DACT1 (mean ± SD of four independent experiments) (d) indicating impaired DVL2 binding of the DACT1 mutants detected here. *p ≤ 0.05; **p ≤ 0.01 (Student’s t test)
Binding of DVL2 to DACT1 mutants is reduced
Human DACT1 interacts with DVL2, a WNT signaling mediator, inducing DVL2 degradation and antagonizing WNT signaling (Zhang et al. 2006). This interaction is mediated by central and C-terminal domains of DACT1, i.e., approximately amino acids 311–836 in the human protein (Zhang et al. 2006; Suriben et al. 2009). Remarkably, 8 of 8 (100%) CAKUT families carry very rare non-silent DACT1 variants that affect amino acids located within the putative DVL2 interaction region of DACT1 (Fig. 1), while this is only the case in 833 of 1,006 (83%) gnomAD controls carrying very rare non-silent DACT1 variants (listed in Supplementary Table 5). To explore the functional consequence of the seven different DACT1 missense variants detected here, we determined DVL2 binding of DACT1 wildtype and mutant proteins in a co-immunoprecipitation assay (Fig. 4c). All seven mutant proteins showed significantly impaired DVL2 binding compared to wildtype DACT1 (Fig. 4d), suggesting that the identified variants act as hypomorphs that may fail to regulate WNT signaling.
Discussion
Based on an index patient with caudal regression syndrome including unilateral kidney agenesis, contralateral duplex kidney with hydronephrosis, anorectal agenesis, sacral dysplasia, and malformations of the central nervous system, this study of 209 families with congenital kidney anomalies associates DACT1 with human CAKUT and characteristic extrarenal features. Very rare non-silent DACT1 variants were significantly more frequent in CAKUT patients of our cohort compared to controls (3.8% versus 1.7%). Moreover, CAKUT patients carrying DACT1 variants were significantly more likely to be additionally affected by anomalies of the digestive or genital tract, skeleton (particularly the spine) or central nervous system, compared to CAKUT patients without DACT1 variants of our cohort. Our data add DACT1 to the list of genes underlying human syndromic CAKUT if mutated. We also establish the kidney and extrarenal phenotype spectrum caused by pathogenic DACT1 variants, more fully defining the spectrum of features in TBS2 (Fig. 5).

Comparison of Townes–Brocks syndrome 1 and 2 according to the literature and this study. a, b Schematic representation of the most prominent phenotypical features of TBS1 caused by heterozygous variants in the SALL1 gene (Kohlhase et al. 1998; Kohlhase 2007) (a) and of TBS2 caused by heterozygous variants in the DACT1 gene (Shi et al. 2012; Nicolaou et al. 2016; Xing et al. 2016; Heidet et al. 2017; Webb et al. 2017; Connaughton et al. 2019 and this study) (b). For detailed case descriptions, see Table 1 and supplementary material including Supplementary Table 4. Please note the phenotypical overlap of both syndromes with respect to features of the central nervous system, eyes, and ears as well as endocrine, heart, skeletal, kidney, gastrointestinal, and genital anomalies. 1Kohlhase et al. (1998); Kohlhase (2007), 2this study (features are given in bold print), 3Shi et al. (2012), 4Webb et al. (2017), 5Connaughton et al. (2019), 6Heidet et al. (2017), 7Nicolaou et al. (2016), 8Xing et al. (2016)
DACT1 (Dapper) is required for notochord formation in Xenopus (Cheyette et al. 2002). Dact1-deficient mice have posterior malformations resembling a human caudal regression syndrome including caudal vertebrae agenesis, anorectal malformation including agenesis, and anomalies of the genitourinary system (Suriben et al. 2009; Wen et al. 2010). Kidney malformations in Dact1-deficient mice included fused kidneys, unilateral or bilateral kidney agenesis, cystic kidneys, and hydronephrosis, the ureters were blind-ended (Suriben et al. 2009; Wen et al. 2010). Except for bilateral kidney agenesis, all of these CAKUT phenotypes were also detected in our patients carrying DACT1 variants, particularly frequently cystic dysplastic kidneys (comprising MCDK, cystic kidney dysplasia, and kidney hypoplasia with a single cyst). Extrarenal anomalies detected in Dact1-deficient mice (Suriben et al. 2009; Wen et al. 2010) and our patients with DACT1 variants included sacral anomalies, anorectal agenesis, genital tract and bladder anomalies, i.e., a spectrum of posterior malformations. With rare exceptions, homozygous Dact1-deficient mice died perinatally (Suriben et al. 2009; Wen et al. 2010), thus psychomotor development, delayed in half of our patients carrying DACT1 variants, could not be monitored. One surviving Dact1-deficient female adult mouse had cystic kidneys, as did more than half of our patients with DACT1 variants, and vaginal agenesis leading to infertility (Wen et al. 2010). Infertility may particularly affect male patients with DACT1 variants, consistent with the finding of blind-ended vas deferens in Dact1-deficient male mice (Wen et al. 2010), because all DACT1 variants identified here were maternally inherited, as far as this could be determined.
The spatiotemporal expression pattern of Dact1 detected here in murine embryos supports a direct role of DACT1 in the development of structures malformed in Dact1-deficient mice and patients with DACT1 variants, such as the vertebrae, anal canal, genital tubercle, kidney and ureter, bladder, and brain. DACT1 inhibits WNT signaling, a function conserved from Xenopus to humans (Cheyette et al. 2002; Zhang et al. 2006). WNT signaling, initiated by binding of extracellular WNT ligands to the transmembrane receptor Frizzled leading to activation of Dishevelled (DVL), plays a key role in embryogenesis and kidney development (Schedl, 2007; Goggolidou 2014; Halt and Vainio 2014; Wang et al. 2018; Meng et al. 2020; https://www.wikipathways.org/index.php/Pathway:WP4150). In kidney organogenesis, WNT-mediated signals control a number of critical processes, such as intermediate mesoderm extension (WNT5A), early ureteric bud branching (WNT11), nephron induction (WNT4 and WNT9B), and the morphogenesis of the medulla (WNT7B) (Schedl 2007; Yu et al. 2009; Halt and Vainio 2014; Yun et al. 2014). In ureter development, WNTs (WNT7B, WNT9B) control smooth muscle differentiation (Trowe et al. 2012). WNT9B, expressed in the epithelial component (ureteric bud), and WNT4, expressed in the metanephric mesenchyme, are encoded by genes that can cause kidney agenesis and hypodysplasia in humans if mutated (Mandel et al. 2008; Vivante et al. 2013; Halt and Vainio 2014; Wu et al. 2017; Lemire et al. 2021; https://www.wikipathways.org/index.php/Pathway:WP5052). Here, we show that DACT1, acting downstream of these WNT ligands, is strongly expressed in the mesenchyme of the ureter and the capsular, cortical, and medullary kidney stroma from murine developmental stage E11.5 to E14.5, is involved in tubulogenesis in vitro and encodes a gene causing CAKUT in humans if mutated. Consistent with our findings that more than half of our patients with DACT1 variants had cystic dysplastic kidneys, WNT signaling is particularly linked to cystic kidney diseases including polycystic kidney disease, nephronophthisis, medullary cystic kidney disease, and HNF1β-associated kidney anomalies (Pulkkinen et al. 2008; Goggolidou 2014). DACT1 antagonizes WNT signaling by binding DVL (Cheyette et al. 2002), and inducing DVL degradation (Zhang et al. 2006), among other mechanisms. Considering that all DACT1 variants identified in this study encode amino acids located within the putative DVL2-binding region (Zhang et al. 2006), and showed impaired DVL2 binding, their inhibitory activity on WNT signaling may be diminished. As DVL is a central component of canonical β-catenin-dependent and non-canonical WNT signaling, both pathways may be affected by DACT1 variants, although DACT1 appears to act mainly upstream of non-canonical planar cell polarity signaling (Suriben et al. 2009; Wen et al. 2010; Yang et al. 2013).
Recently, a heterozygous DACT1 nonsense variant in a three-generation family was proposed to cause a TBS-like syndrome referred to as TBS2 (Webb et al. 2017). Autosomal dominant TBS1 is characterized by the triad of imperforate anus or anal stenosis in 84%, dysplastic ears in 87%, and thumb malformations in 89%, and is caused by variants in the SALL1 (spalt like transcription factor 1) gene (Kohlhase et al. 1998; Kohlhase 2007). Functional kidney impairment with or without structural abnormalities, including polycystic kidneys, has been reported in 42% of individuals with TBS1 (Kohlhase 2007), and rare SALL1 variants were detected in 0.5–1.4% of CAKUT patients (Hwang et al. 2014; Heidet et al. 2017; Kosfeld et al. 2018). Several anomalies in the family with TBS2 carrying a pathogenic DACT1 variant (Webb et al. 2017) overlap with TBS1 (Kohlhase et al. 1998; Kohlhase 2007), affecting the central nervous system, the ears, the endocrine system, the kidneys, the gastrointestinal and genital tract, and the skeleton, whereas thumb abnormalities were not observed (Fig. 5). Here, we report eight new families with heterozygous DACT1 variants, emphasizing the importance of kidney anomalies and supporting the observation of a characteristic phenotype spectrum additionally involving the skeleton (particularly the spine), the digestive and genital tract, and the central nervous system in TBS2, similar to that in TBS1 (Fig. 5). This combination of phenotypic features in TBS2 is confirmed when combining our data with that from the literature totaling 26 patients from 19 families with very rare DACT1 variants, investigated because of neural tube defects (Shi et al. 2012), Müllerian duct (Xing et al. 2016) or kidney anomalies (Nicolaou et al. 2016; Heidet et al. 2017; Connaughton et al. 2019; this study) or TBS-like features (Webb et al. 2017). Families carrying DACT1 variants recurrently presented with anomalies of the kidney (12/19, 63%), skeleton (12/19, 63%), central nervous system (10/19, 53%), genital tract (4/19, 21%), lung (3/19, 16%), and distal digestive tract (2/19, 10.5%) (Fig. 5; Supplementary Table 4). Of note, congenital kidney anomalies were observed in almost two-thirds of families. Thus, similar to the mouse model, in which Dact1 deficiency is fully penetrant for kidney anomalies (Suriben et al. 2009; Wen et al. 2010), CAKUT seem to be a major feature in patients with rare heterozygous DACT1 variants.
However, incomplete penetrance with respect to CAKUT and extrarenal phenotypes and variable expressivity is observed in individuals carrying heterozygous DACT1 variants that may be unaffected or show variable features of TBS2, leading to miscarriage in the worst case (Fig. 1, Supplementary Table 4). Similarly, few Dact1-deficient mice survive postnatally and present with milder malformations, while most die perinatally due to severe developmental defects (Suriben et al. 2009; Wen et al. 2010). Incomplete penetrance is commonly observed in autosomal dominant familial CAKUT, and environmental factors or epigenetic alterations may contribute to CAKUT pathogenesis and severity of defects (Sanna-Cherchi et al. 2018; van der Ven et al. 2018b; Nigam et al. 2019). Similar to findings in patients with heterozygous DACT1 variants, heterozygous variants in SALL1 have been identified in patients with typical features of TBS1, but also in patients with isolated CAKUT (Hwang et al. 2014; Heidet et al. 2017; Kosfeld et al. 2018).
In conclusion, we identified very rare heterozygous missense variants in the DVL2 interaction region of DACT1 in 3.8% of CAKUT families. We provide further evidence that deleterious DACT1 variants and Dact1 deficiency cause kidney anomalies and a defined spectrum of extrarenal malformations in mice and humans, referred to as TBS2. When identified in CAKUT patients with features of TBS2, rare DACT1 variants may be considered causative, especially in cases without SALL1 variants. Kidney ultrasound is warranted in patients carrying rare DACT1 variants since two-thirds of families described so far present with kidney agenesis, duplex/fused or (multi)cystic (hypo)dysplastic kidneys with hydronephrosis.
References
Brophy PD, Rasmussen M, Parida M et al (2017) A gene implicated in activation of retinoic acid receptor targets is a novel renal agenesis gene in humans. Genetics 207:215–228. https://doi.org/10.1534/genetics.117.1125
Chen D, Roberts R, Pohl M et al (2004) Differential expression of collagen- and laminin-binding integrins mediates ureteric bud and inner medullary collecting duct cell tubulogenesis. Am J Physiol Renal Physiol 287:F602–F611. https://doi.org/10.1152/ajprenal.00015.2004
Cheyette BN, Waxman JS, Miller JR et al (2002) Dapper, a Dishevelled-associated antagonist of beta-catenin and JNK signaling, is required for notochord formation. Dev Cell 2:449–461. https://doi.org/10.1016/s1534-5807(02)00140-5
Christians A, Weiss AC, Martens H et al (2020) Inflammation-like changes in the urothelium of Lifr-deficient mice and LIFR-haploinsufficient humans with urinary tract anomalies. Hum Mol Genet 29:1192–1204. https://doi.org/10.1093/hmg/ddaa048
Connaughton DM, Kennedy C, Shril S et al (2019) Monogenic causes of chronic kidney disease in adults. Kidney Int 95:914–928. https://doi.org/10.1016/j.kint.2018.10.031
Connaughton DM, Dai R, Owen DJ et al (2020) Mutations of the transcriptional corepressor ZMYM2 cause syndromic urinary tract malformations. Am J Hum Genet 107:727–742. https://doi.org/10.1016/j.ajhg.2020.08.013
De Tomasi L, David P, Humbert C et al (2017) Mutations in GREB1L cause bilateral kidney agenesis in humans and mice. Am J Hum Genet 101:803–814. https://doi.org/10.1016/j.ajhg.2017.09.026
Goggolidou P (2014) Wnt and planar cell polarity signaling in cystic renal disease. Organogenesis 10:86–95. https://doi.org/10.4161/org.26766
Halt K, Vainio S (2014) Coordination of kidney organogenesis by Wnt signaling. Pediatr Nephrol 29:737–744. https://doi.org/10.1007/s00467-013-2733-z
Harambat J, van Stralen KJ, Kim JJ et al (2012) Epidemiology of chronic kidney disease in children. Pediatr Nephrol 27:363–373. https://doi.org/10.1007/s00467-011-1939-1
Heidet L, Moriniere V, Henry C et al (2017) Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 28:2901–2914. https://doi.org/10.1681/ASN.2017010043
Humbert C, Silbermann F, Morar B et al (2014) Integrin alpha 8 recessive mutations are responsible for bilateral renal agenesis in humans. Am J Hum Genet 94:288–294. https://doi.org/10.1016/j.ajhg.2013.12.017
Hwang DY, Dworschak GC, Kohl S et al (2014) Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int 85:1429–1433. https://doi.org/10.1038/ki.2013.508
Kircher M, Witten DM, Jain P et al (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. https://doi.org/10.1038/ng.2892
Kohlhase J, Wischermann A, Reichenbach H et al (1998) Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet 18:81–83. https://doi.org/10.1038/ng0198-81
Kohlhase J (2007) Townes–Brocks syndrome. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A (eds) GeneReviews®. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK1445/
Kosfeld A, Kreuzer M, Daniel C et al (2016) Whole-exome sequencing identifies mutations of TBC1D1 encoding a Rab-GTPase-activating protein in patients with congenital anomalies of the kidneys and urinary tract (CAKUT). Hum Genet 135:69–87. https://doi.org/10.1007/s00439-015-1610-1
Kosfeld A, Brand F, Weiss AC et al (2017) Mutations in the leukemia inhibitory factor receptor (LIFR) gene and Lifr deficiency cause urinary tract malformations. Hum Mol Genet 26:1716–1731. https://doi.org/10.1093/hmg/ddx086
Kosfeld A, Martens H, Hennies I et al (2018) Kongenitale Anomalien der Nieren und ableitenden Harnwege (CAKUT). medizinische genetik 30:448–460. https://doi.org/10.1007/s11825-018-0226-y
Lemire G, Zheng B, Ediae GU et al (2021) Homozygous WNT9B variants in two families with bilateral renal agenesis/hypoplasia/dysplasia. Am J Med Genet A 185:3005–3011. https://doi.org/10.1002/ajmg.a.62398
Limwongse C (2009) Syndromes and malformations of the urinary tract. In: Avner E, Harmon W, Niaudet P, Yoshikawa N (eds) Pediatric Nephrology. Springer, Berlin, pp 121–156
Mai W, Chen D, Ding T et al (2005) Inhibition of Pkhd1 impairs tubulomorphogenesis of cultured IMCD cells. Mol Biol Cell 16:4398–4409. https://doi.org/10.1091/mbc.e04-11-1019
Mandel H, Shemer R, Borochowitz ZU et al (2008) SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet 82:39–47. https://doi.org/10.1016/j.ajhg.2007.08.005
Martens H, Hennies I, Getwan M et al (2020) Rare heterozygous GDF6 variants in patients with renal anomalies. Eur J Hum Genet 28:1681–1693. https://doi.org/10.1038/s41431-020-0678-9
Meng P, Zhu M, Ling X et al (2020) Wnt signaling in kidney: the initiator or terminator? J Mol Med 98:1511–1523. https://doi.org/10.1007/s00109-020-01978-9
Moorman AF, Houweling AC, de Boer PA et al (2001) Sensitive nonradioactive detection of mRNA in tissue sections: Novel application of the whole-mount in situ hybridization protocol. J Histochem Cytochem 49:1–8. https://doi.org/10.1177/002215540104900101
Münch J, Engesser M, Schönauer R et al (2022) Biallelic pathogenic variants in roundabout guidance receptor 1 associate with syndromic congenital anomalies of the kidney and urinary tract. Kidney Int 101:1039–1053. https://doi.org/10.1016/j.kint.2022.01.028
Nicolaou N, Pulit SL, Nijman IJ et al (2016) Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int 89:476–486. https://doi.org/10.1038/ki.2015.319
Nigam A, Knoers N, Renkema KY (2019) Impact of next generation sequencing on our understanding of CAKUT. Semin Cell Dev Biol 91:104–110. https://doi.org/10.1016/j.semcdb.2018.08.013
Pohl M, Bhatnagar V, Mendoza SA et al (2002) Toward an etiological classification of developmental disorders of the kidney and upper urinary tract. Kidney Int 61:10–19. https://doi.org/10.1046/j.1523-1755.2002.00086.x
Pulkkinen K, Murugan S, Vainio S (2008) Wnt signaling in kidney development and disease. Organogenesis 4:55–59. https://doi.org/10.4161/org.4.2.5849
Queisser-Luft A, Stolz G, Wiesel A et al (2002) Malformations in newborn: results based on 30,940 infants and fetuses from the Mainz congenital birth defect monitoring system (1990–1998). Arch Gynecol Obstet 266:163–167. https://doi.org/10.1007/s00404-001-0265-4
Ran FA, Hsu PD, Wright J et al (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. https://doi.org/10.1038/nprot.2013.143
Rentzsch P, Witten D, Cooper GM et al (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47:D886–D894. https://doi.org/10.1093/nar/gky1016
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Sanna-Cherchi S, Sampogna RV, Papeta N et al (2013) Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med 369:621–629. https://doi.org/10.1056/NEJMoa1214479
Sanna-Cherchi S, Westland R, Ghiggeri GM et al (2018) Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 128:4–15. https://doi.org/10.1172/JCI95300
Schedl A (2007) Renal abnormalities and their developmental origin. Nat Rev Genet 8:791–802. https://doi.org/10.1038/nrg2205
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. https://doi.org/10.1038/nmeth.2089
Shi Y, Ding Y, Lei YP et al (2012) Identification of novel rare mutations of DACT1 in human neural tube defects. Hum Mutat 33:1450–1455. https://doi.org/10.1002/humu.22121
Stoll C, Dott B, Alembik Y et al (2014) Associated nonurinary congenital anomalies among infants with congenital anomalies of kidney and urinary tract (CAKUT). Eur J Med Genet 57:322–328. https://doi.org/10.1016/j.ejmg.2014.04.014
Suriben R, Kivimae S, Fisher DA et al (2009) Posterior malformations in Dact1 mutant mice arise through misregulated Vangl2 at the primitive streak. Nat Genet 41:977–985. https://doi.org/10.1038/ng.435
Trowe MO, Airik R, Weiss AC et al (2012) Canonical Wnt signaling regulates smooth muscle precursor development in the mouse ureter. Development 139:3099–3108. https://doi.org/10.1242/dev.077388
van der Ven AT, Connaughton DM, Ityel H et al (2018a) Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 29:2348–2361. https://doi.org/10.1681/ASN.2017121265
van der Ven AT, Vivante A, Hildebrandt F (2018b) Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 29:36–50. https://doi.org/10.1681/ASN.2017050561
Verbitsky M, Westland R, Perez A et al (2019) The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet 51:117–127. https://doi.org/10.1038/s41588-018-0281-y
Vivante A, Mark-Danieli M, Davidovits M et al (2013) Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J Am Soc Nephrol 24:550–558. https://doi.org/10.1681/ASN.2012010097
Vivante A, Kleppa MJ, Schulz J et al (2015) Mutations in TBX18 cause dominant urinary tract malformations via transcriptional dysregulation of ureter development. Am J Hum Genet 97:291–301. https://doi.org/10.1016/j.ajhg.2015.07.001
Wang Y, Zhou CJ, Liu Y (2018) Wnt signaling in kidney development and disease. Prog Mol Biol Transl Sci 153:181–207. https://doi.org/10.1016/bs.pmbts.2017.11.019
Webb BD, Metikala S, Wheeler PG et al (2017) Heterozygous pathogenic variant in DACT1 causes an autosomal-dominant syndrome with features overlapping townes-brocks syndrome. Hum Mutat 38:373–377. https://doi.org/10.1002/humu.23171
Weber S, Landwehr C, Renkert M et al (2011) Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrol Dial Transplant 26:136–143. https://doi.org/10.1093/ndt/gfq400
Wen J, Chiang YJ, Gao C et al (2010) Loss of Dact1 disrupts planar cell polarity signaling by altering dishevelled activity and leads to posterior malformation in mice. J Biol Chem 285:11023–11030. https://doi.org/10.1074/jbc.M109.085381
Wu H, Xu Q, Xie J et al (2017) Identification of 8 novel mutations in nephrogenesis-related genes in chinese han patients with unilateral renal agenesis. Am J Nephrol 46:55–63. https://doi.org/10.1159/000477590
Xing Q, Xu Z, Zhu Y et al (2016) Genetic analysis of DACT1 in 100 Chinese Han women with Mullerian duct anomalies. Reprod Biomed Online 32:420–426. https://doi.org/10.1016/j.rbmo.2016.01.003
Yang X, Fisher DA, Cheyette BN (2013) SEC14 and spectrin domains 1 (Sestd1), dishevelled 2 (Dvl2) and dapper antagonist of catenin-1 (Dact1) co-regulate the Wnt/planar cell polarity (PCP) pathway during mammalian development. Commun Integr Biol 6:e26834. https://doi.org/10.4161/cib.26834
Yang N, Wu N, Dong S et al (2020) Human and mouse studies establish TBX6 in Mendelian CAKUT and as a potential driver of kidney defects associated with the 16p11.2 microdeletion syndrome. Kidney Int 98:1020–1030. https://doi.org/10.1016/j.kint.2020.04.045
Yu J, Carroll TJ, Rajagopal J et al (2009) A Wnt7b-dependent pathway regulates the orientation of epithelial cell division and establishes the cortico-medullary axis of the mammalian kidney. Development 136:161–171. https://doi.org/10.1242/dev.02208
Yun K, Ajima R, Sharma N et al (2014) Non-canonical Wnt5a/Ror2 signaling regulates kidney morphogenesis by controlling intermediate mesoderm extension. Hum Mol Genet 23:6807–6814. https://doi.org/10.1093/hmg/ddu397
Zhang L, Gao X, Wen J et al (2006) Dapper 1 antagonizes Wnt signaling by promoting dishevelled degradation. J Biol Chem 281:8607–8612. https://doi.org/10.1074/jbc.M600274200
Acknowledgements
The authors wish to thank the patients and their families for participating in this study, Achim Gossler (Institute of Molecular Biology, Hannover Medical School, Hannover, Germany) for providing wildtype mIMCD3 cells, and the Cell Sorting Core Facility, Hannover Medical School, Hannover, Germany for technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to Anne Christians [KO5614/2-1] and Helge Martens [MA9606/1-1].
Author information
Authors and Affiliations
Contributions
AC and RGW designed the study and conceived the experiments. AC, EK, M-OT, and FB carried out the experiments. RG generated the exome sequencing raw data. IH, AH, ACG, ZG, MZ, TS, HB, AB, VT, BU, DH, and JD contributed patient material, clinical information, and performed reverse phenotyping. AC, EK, M-OT, FB, HM, AK, and RGW analyzed the data. AC, EK, and HM generated the figures. AC and RGW wrote the manuscript with contributions from all authors. All authors approved the submitted and published version of the manuscript. This study includes thesis work of EK (MSc and Dr. rer. nat.).
Corresponding author
Ethics declarations
Conflicts of interests
The authors declare that they have no conflicts of interests.
Ethics approval
This study was conducted according to the World Medical Association (WMA) Declaration of Helsinki (64th WMA General Assembly, Fortaleza, Brazil, October 2013). Written informed consent from the parents of all patients was received prior to inclusion in this study. All applicable international, national and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Christians, A., Kesdiren, E., Hennies, I. et al. Heterozygous variants in the DVL2 interaction region of DACT1 cause CAKUT and features of Townes–Brocks syndrome 2. Hum Genet 142, 73–88 (2023). https://doi.org/10.1007/s00439-022-02481-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-022-02481-6