
Abstract
An estimated 5–11% of patients with neurofibromatosis type-1 (NF1) harbour large deletions encompassing the NF1 gene and flanking regions. These NF1 microdeletions are subclassified into type 1, 2, 3 and atypical deletions which are distinguishable from each other by their extent and by the number of genes included within the deletion regions as well as the frequency of mosaicism with normal cells. Most common are type-1 NF1 deletions which encompass 1.4-Mb and 14 protein-coding genes. Type-1 deletions are frequently associated with overgrowth, global developmental delay, cognitive disability and dysmorphic facial features which are uncommon in patients with intragenic pathogenic NF1 gene variants. Further, patients with type-1 NF1 deletions frequently exhibit high numbers of neurofibromas and have an increased risk of malignant peripheral nerve sheath tumours. Genes located within the type-1 NF1 microdeletion interval and co-deleted with NF1 are likely to act as modifiers responsible for the severe disease phenotype in patients with NF1 microdeletions, thereby causing the NF1 microdeletion syndrome. Genotype/phenotype correlations in patients with NF1 microdeletions of different lengths are important to identify such modifier genes. However, these correlations are critically dependent upon the accurate characterization of the deletions in terms of their extent. In this review, we outline the utility as well as the shortcomings of multiplex ligation-dependent probe amplification (MLPA) to classify the different types of NF1 microdeletion and indicate the importance of high-resolution microarray analysis for correct classification, a necessary precondition to identify those genes responsible for the NF1 microdeletion syndrome.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Neurofibromatosis type 1 (NF1; MIM#162200) is one of the most common inherited cancer predisposition syndromes with an estimated frequency of 1:3000 (Lammert et al. 2005). Among all patients with NF1, 5–11% of patients have large deletions encompassing the entire NF1 gene and its flanking regions at 17q11.2 (Cnossen et al. 1997; Rasmussen et al. 1998; Kluwe et al. 2004; Pasmant et al. 2015; Zhang et al. 2015). These ‘NF1 microdeletions’ are often associated with a severe clinical manifestation of NF1 causing the NF1 microdeletion syndrome (MIM#613576). Considered as a group, NF1 microdeletion patients often have a more severe form of NF1 as compared to patients with intragenic pathogenic NF1 variants. However, a certain degree of variability in terms of clinical symptoms has been observed on an individual level when comparing different patients with NF1 microdeletions. These clinical phenotypic differences are likely to be caused by various factors including differences in deletion size and hence the number of genes co-deleted with the NF1 gene. Four types of large NF1 deletion (type-1, 2, 3 and atypical) have been identified that are distinguishable in terms of their size and breakpoint location, by the number of genes located within the deletion region and by the frequency of somatic mosaicism with normal cells lacking the deletion. Somatic mosaicism with normal cells is likely to cause a milder disease manifestation in patients with NF1 microdeletions as compared to patients with germline NF1 microdeletions.
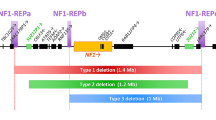
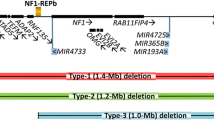
Most frequent among all NF1 microdeletions are the type-1 NF1 deletions which encompass 1.4-Mb and include 14 protein-coding genes as well as five microRNA genes (Fig. 1) (Dorschner et al. 2000; Jenne et al. 2001; López-Correa et al. 2001). Type-1 deletions account for 70–80% of all large NF1 deletions and usually occur as germline lesions that are present in all cells of the affected patients (Messiaen et al. 2011; Summerer et al. 2018). Most type-1 NF1 deletions are caused by interchromosomal non-allelic homologous recombination (NAHR) during maternal meiosis (López-Correa et al. 2000; Neuhäusler et al. 2018). The NAHR events causing type-1 NF1 deletions are mediated by the low-copy repeats, NF1-REPa and NF1-REPc. Within these low-copy repeats, recurrent breakpoints have been detected within two NAHR hotspots, termed paralogous recombination sites 1 and 2 (PRS1 and PRS2) (Forbes et al. 2004; De Raedt et al. 2006; Bengesser et al. 2014; Hillmer et al. 2016, 2017).

Schema of the 1.4-Mb spanning type-1 NF1 microdeletion region and its flanking regions indicating the relative positions of the 14 protein-coding genes, the SUZ12P pseudogene and the 5 microRNA genes located there. The relative extent of type-1, type-2, atypical deletions of group #2A and type-3 NF1 deletions are indicated by grey horizontal bars. The vertical red and green arrows represent the binding sites of the MLPA-probes included in the SALSA® MLPA® Probemix P122-D2 NF1-area (MRC-Holland). Red arrows represent MLPA-probes targeting genomic regions encompassed by the respective deletions whereas green arrows represent MLPA-probes targeted to regions which are not deleted and present in two copies. For example, in case of type-1 NF1 deletions encompassing 1.4-Mb, the target sequences for the probes shown in red are present in only one copy because they are located within the deletion region. By contrast, the target sequences for the MLPA-probes shown in green are present in two copies since they are not located within the deletion region. The MLPA-probe SUZ12P intron 4 is shaded in green and red because the region targeted by this probe is deleted in some but not all type-2 and atypical group #2A NF1 deletions. Type-2 deletions and atypical group #2A deletions cannot be distinguished by means of MLPA. cen centromeric; tel telomeric
In contrast to type-1 NF1 deletions, type-2 deletions encompass only 1.2-Mb and are associated with hemizygosity for 13 protein-coding genes since the LRRC37B gene is not located within the type-2 NF1 deletion interval (Fig. 1). It has been estimated that 10% of all NF1 microdeletions are type-2 (Messiaen et al. 2011). NAHR between the SUZ12 gene and its pseudogene SUZ12P is the cause of the vast majority of type-2 NF1 deletions (Vogt et al. 2012). In contrast to type-1 deletions, type-2 NF1 deletions are frequently of postzygotic origin, mediated by mitotic NAHR, and hence are associated with somatic mosaicism of normal cells without the deletion (Kehrer-Sawatzki et al. 2004; Steinmann et al. 2007; Roehl et al. 2010, 2012). It has been estimated that at least 70% of all type-2 deletions are mosaic with high numbers of cells (94–99%) harbouring the deletion in blood, but with lower proportions in skin fibroblasts (39–91%) and urine cells (24–82%) (Steinmann et al. 2007; Messiaen et al. 2011; Roehl et al. 2012). Type-3 NF1 deletions are rare, occurring in only 1–4% of all patients with NF1 microdeletions. They encompass 1-Mb and are mediated by NAHR between NF1-REPb and NF1-REPc leading to hemizygosity for a total of 9 protein-coding genes (Fig. 1) (Bengesser et al. 2010; Pasmant et al. 2010; Zickler et al. 2012).
In contrast to type-1, 2 and 3 NF1 deletions, atypical NF1 deletions do not have recurrent breakpoints and are heterogeneous in terms of their size and the number of genes located within the deleted region (reviewed by Kehrer-Sawatzki et al. submitted for publication). Approximately 8–10% of all NF1 microdeletions are considered to be atypical (Pasmant et al. 2010; Messiaen et al. 2011). They may occur as germline deletions but can also be of postzygotic origin and hence may be associated with somatic mosaicism with normal cells (Taylor Tovares et al. 2013). It has been estimated that 59% of atypical NF1 deletions are of postzygotic origin and thus represent mosaic deletions (Vogt et al. 2014). Atypical NF1 deletions are caused by a multitude of mutational mechanisms including aberrant DNA double strand break repair, replication-associated errors and retrotransposon-mediated mechanisms (Vogt et al. 2014 and references therein).
Patients with NF1 microdeletions often exhibit more severe clinical manifestations of NF1 than patients with intragenic pathogenic NF1 variants and their clinical phenotype has been mainly investigated in patients with type-1 NF1 deletions (Pasmant et al. 2010; Mautner et al. 2010; Pacot et al. 2021; reviewed by Kehrer-Sawatzki et al. 2017). The lifetime risk of a malignant peripheral nerve sheath tumour (MPNST) in patients with type-1 NF1 deletions is in the range of 16–26% (De Raedt et al. 2003; Mautner et al. 2010) which is higher than the estimated lifetime risk for an MPNST in all NF1 patients which is 8–15.8% (Evans et al. 2002, 2012; Uusitalo et al. 2016). Further, MPNSTs may occur significantly earlier in patients with NF1 microdeletions as compared with NF1 patients with intragenic pathogenic variants (De Raedt et al. 2003). Higher numbers of subcutaneous and plexiform neurofibromas and higher growth rates of these tumours have been observed in patients with NF1 microdeletions as compared to patients with intragenic pathogenic NF1 variants (Well et al. 2021). In addition, many patients with NF1 microdeletions exhibit features which are not usually observed in patients with pathogenic variants within the NF1 gene including facial dysmorphic features, overgrowth, severe global developmental delay and intellectual disability (reviewed by Kehrer-Sawatzki et al. 2017, 2020; Ottenhoff et al. 2020).
It has been postulated that some of the genes co-deleted with NF1 exert an influence on the clinical manifestation of the disease in patients with NF1 microdeletions. However, to derive accurate and reliable genotype/phenotype correlations, a precise classification of the types of NF1 microdeletion is very important since the different types of deletion differ in terms of their extent and hence the number of genes encompassed by the deletion.
In the past, multiplex ligation-dependent probe amplification (MLPA) has been routinely used to identify and characterize NF1 microdeletions (Wimmer et al. 2006; De Luca et al. 2007). However, there are limitations with regard to the accurate classification of NF1 microdeletions when MLPA is employed as the only method of analysis. In this review, we outline the utility as well as the limitations of MLPA in relation to the classification of NF1 microdeletions and discuss alternative methods that may be employed to classify these gross deletions. This is important in the context of establishing genotype/phenotype correlations in patients with NF1 microdeletions since the different types of NF1 deletion are associated with the loss of a variable number of genes.
Classification of NF1 microdeletions by MLPA
The SALSA® MLPA® Probemix P122-D2 NF1-area (MRC-Holland) includes 35 MLPA-probes. Ten of these are reference probes that detect autosomal regions not located on chromosome 17. Additionally, the probemix contains 23 MLPA-probes that are designed to detect regions located within the chromosomal region 17q11.2 (Table 1). Of the 23 MLPA-probes, 14 map to the type-1 NF1 microdeletion region (Fig. 1). The probemix also contains two MLPA-probes that map to the short arm of chromosome 17 (17p11.2). A dosage quotient between 0.40 and 0.65 for any given probe is considered to be indicative of a hemizygous deletion. The SALSA® MLPA® Probemix P122-D2 NF1-area is the only commercially available test to identify NF1 microdeletions by MLPA and it is widely used for clinical diagnostic purposes.
In the following, both the utility and the accuracy of the MLPA® Probemix P122-D2 NF1-area to classify NF1 deletions are outlined for each type of deletion.
Type-1 NF1 microdeletions
If the target sequences of the 14 MLPA probes shown in red in Fig. 1 are present in only one copy in a patient’s DNA, the NF1 deletion is considered to be of type-1. Typical MLPA results indicative of a type-1 NF1 deletion are presented in Table 1. Type-1 NF1 deletions encompass the region targeted by probe MLPA-probe LRRC37B which maps to exon 1 of the LRRC37B gene located within NF1-REPc. The genomic region targeted by both MLPA-probes for the SUZ12P pseudogene is present in only a single copy in the case of a type-1 NF1 deletion (Table 1). The MLPA results given in Table 1 are typical for type-1 NF1 deletions and not for any other type of NF1 microdeletion. There are, however, considerable distances (269 kb and 345 kb) between the MLPA-probes immediately flanking the type-1 deletion breakpoint regions located within NF1-REPa and NF1-REPc (Fig. 1). In view of the large distances between these MLPA-probes, it may be argued that MLPA is not on its own precise enough to unambiguously classify type-1 NF1 deletions on the grounds that the deletion breakpoints may not be located within NF1-REPa and NF1-REPc as is characteristic for type-1 deletions. To analyse this in greater detail, Summerer et al. (2018) investigated 236 unselected NF1 microdeletions which were initially identified by MLPA and presumed to be of type-1 since they encompassed the region targeted by MLPA-probe LRRC37B and both MLPA-probes for the SUZ12P pseudogene, as indicated in Table 1. Summerer et al. (2018) performed custom-designed array CGH (Agilent SurePrint G3 human CGH microarray) to improve the breakpoint prediction of type-1 NF1 deletions as well as CytoScan ™ HD array analysis (Affymetrix). The authors precisely identified the deletion breakpoint regions by sequencing breakpoint-spanning PCR products thereby determining the breakpoints at the highest possible resolution. Of the 236 deletions investigated, 234 (99.2%) were classified as bona fide type-1 NF1 deletions. All 234 deletions had breakpoints located within NF1-REPa and NF1-REPc and were mediated by NAHR, the main mechanism underlying type-1 NF1 deletions. Only in two of the 236 deletions could the breakpoints not be identified by breakpoint-spanning PCR. Nevertheless, the results of the microarray analysis indicated that the breakpoints of these two deletions were also located within NF1-REPa and NF1-REPc (Summerer et al. 2018). Hence, their analysis of the 236 deletions indicated that NF1 deletions demarcated by MLPA (as indicated in Table 1) are highly likely to be of type-1. MLPA therefore represents a very efficient method with which to identify type-1 NF1 deletions. Microarray analysis applied in addition to MLPA could be used to confirm that the deletion breakpoints are indeed located with NF1-REPa and NF-REPc, as is characteristic of type-1 NF1 deletions. This may be relevant in patients who present with an unusual clinical phenotype which differs from that seen in the majority of patients with type-1 NF1 deletions.
If patients with type-1 NF1 deletions present with additional clinical symptoms not frequently encountered in patients with NF1 microdeletions, the possibility should be considered that additional pathogenic variants might be present in unlinked genes, as recently reported by Santorro et al. (2021). These authors reported a male patient with a type-1 NF1 deletion and clinical features of the NF1 microdeletion syndrome that were complicated by cleft palate and other dysmorphic features, hypoplasia of corpus callosum, and partial bicoronal craniosynostosis caused by a novel 2 bp deletion in exon 2 of the Meis homeobox 2 gene (MEIS2) inherited from his mildly affected father.
It is important to emphasize that, even using microarray analysis, the breakpoints within NF1-REPa and NF1-REPb cannot be unambiguously assigned owing to the highly repetitive nature of the sequences within the NF1-REPs. Sequences with high homology to the NF1-REPs are present in multiple copies on chromosome 17 (Giannuzzi et al. 2013). Owing to the repetitivity of these sequences, the breakpoints cannot be precisely detected by microarray analysis with standard software tools used to analyse germline chromosomal aberrations (Summerer et al. 2018). The breakpoints of type-1 deletions can only be narrowed down by means of breakpoint-spanning PCRs and sequence analysis of the respective PCR products (Hillmer et al. 2017; Summerer et al. 2018). Of the 236 type-1 NF1 deletions analysed by Summerer et al. (2018), 179 (75.8%) harboured breakpoints within the NAHR hotspot PRS2 which spans 4.8 kb. By contrast, 39 (16.5%) type-1 deletions had breakpoints within PRS1 encompassing 5.2 kb. Most of the remaining 18 deletions exhibited breakpoints that were located between PRS1 and PRS2 within a genomic region of 14 kb exhibiting high sequence similarity between NF1-REPa and NF1-REPc. In total, 13 (5.5%) of the 236 type-1 deletions analysed had breakpoints within this 14 kb region (Summerer et al. 2018).
Type-3 NF1 deletions
Only 8 type-3 NF1 deletions have been identified so far by means of accurate breakpoint analysis (Bengesser et al. 2010; Pasmant et al. 2010; Zickler et al. 2012). All of them exhibited the same MLPA results as indicated in Table 1. By means of deletion breakpoint-spanning PCR and sequence analysis of the PCR-products, the breakpoints of these 8 type-3 NF1 deletions were identified within homologous regions between NF1-REPb and NF1-REPc indicative of NAHR as the underlying mechanism. It follows that type-3 NF1 deletions ascertained using MLPA, as indicated in Table 1, are likely to be bona fide. However, owing to the large distances between the MLPA-probes flanking the breakpoint regions, additional tools, such as breakpoint-spanning PCRs or microarray analysis, are required in order to finally confirm the presence of a bona fide type-3 deletion.
Zhang et al. (2015) identified a large NF1 deletion in patient NF073 with the same MLPA-probe pattern as that observed for the 8 type-3 NF1 deletions mentioned above. The deletion of patient NF073 was classified as atypical by Zhang et al. (2015). However, since MLPA was the only analytical method performed, an unambiguous distinction between type-3 and atypical deletion could not be made. To distinguish between both types of NF1 deletion would be important in terms of assessing the number of genes encompassed by the deletion, the likelihood of somatic mosaicism with normal cells, as well as the mutational mechanism underlying the corresponding deletion.
Type-2 NF1 deletions
Type-2 deletions cannot be classified with any degree of accuracy by employing MLPA as the single method of analysis. This conclusion may be drawn from the findings of Vogt et al. (2012, 2014) who analysed type-2 as well as atypical NF1 deletions and compared the corresponding MLPA results. In all, Vogt et al. analysed 40 type-2 NF1 deletions with breakpoints located within SUZ12 and SUZ12P as confirmed by breakpoint-spanning PCRs. A breakpoint localization within SUZ12 and SUZ12P is characteristic of type-2 deletions. The 40 type-2 deletions analysed by Vogt et al. (2012) exhibited breakpoints within sequences homologous between SUZ12 and SUZ12P, which is indicative of NAHR being the major causative mechanism underlying these deletions (Vogt et al. 2012). Two different MLPA results were obtained for these 40 type-2 NF1 deletions (Table 2). Nine of the 40 deletions encompassed the region corresponding to MLPA-probe SUZ12P intron 4. By contrast, 31 of the 40 type-2 NF1 deletions did not include the region corresponding to this MLPA-probe (Table 2). None of the 40 type-2 deletions analysed by Vogt et al. (2012) encompassed the region targeted by MLPA-probe SUZ12 exon 10, which is not located within the region of sequence homology between SUZ12 and SUZ12P.
However, the MLPA results observed in the 40 type-2 NF1 deletions analysed by Vogt et al. (2012) are by no means exclusive to type-2 NF1 deletions. Vogt et al. (2012, 2014) identified 9 of 19 atypical NF1 deletions exhibiting the same MLPA results as those observed for type-2 NF1 deletions. As determined by breakpoint-spanning PCR or custom-designed MLPA, the breakpoints of these 9 atypical NF1 deletions were not located within homologous regions between SUZ12 and SUZ12P as is characteristic for type-2 deletions. Instead, the proximal breakpoints of these 9 deletions were located either within SUZ12P, CRLF3 or between SUZ12P and CRLF3. The telomeric deletion breakpoints were located either within the UPT6 gene or the genomic regions between UTP6 and SUZ12 (Table 3). The molecular mechanism responsible for these atypical NF1 deletions was not NAHR but instead non-homologous end joining (NHEJ) or a replication-based mechanism (Vogt et al. 2014). In the following, these atypical NF1 deletions are termed group #2A deletions.
According to the findings of Vogt et al. (2012, 2014), type-2 NF1 deletions and atypical group #2A deletions cannot be distinguished from one another by MLPA. Other techniques such as microarray analysis, specifically tailored to detect chromosomal aberrations at high resolution [as performed by Pasmant et al. (2009), Vogt et al. (2014) and Summerer et al. (2019)] or sequence analysis of breakpoint-spanning PCRs, have to be employed to distinguish between these types of NF1 deletion. This may well be of clinical significance since SUZ12 is functionally inactivated by the breakpoints of type-2 deletions. SUZ12 inactivation by intragenic pathogenic variants has been shown to cause overgrowth, dysmorphic features, musculoskeletal abnormalities and developmental delay/intellectual disability (Imagawa et al. 2018; Cyrus et al. 2019a, b). By contrast, SUZ12 is present in two copies and is not affected by the breakpoints of atypical NF1 deletions of group #2A.
NAHR between SUZ12 and SUZ12P is the major mechanism underlying type-2 NF1 deletions. However, rare cases of type-2 deletions have been reported that exhibit breakpoints at non-homologous sites within SUZ12 and SUZ12P and hence are not mediated by NAHR (Vogt et al. 2012). These deletions could not be distinguished from type-2 deletions mediated by NAHR if MLPA using the SALSA® MLPA® Probemix P122-D2 NF1-area were the only analytical method employed to characterize these deletions. Microarray-analysis using targeted arrays, custom-designed MLPA analysis and breakpoint-spanning PCRs would be necessary to narrow down the breakpoints of type-2 deletions and distinguish between those mediated by NAHR and those caused by other mutational mechanisms (Vogt et al. 2012).
Atypical NF1 deletions
As yet, a total of 61 atypical NF1 deletions have been reported in the literature (reviewed by Kehrer-Sawatzki et al. submitted for publication). In contrast to the NF1 deletions mediated by NAHR, atypical NF1 deletions do not exhibit recurrent breakpoints and are quite heterogeneous in terms of their size and the number of genes located within the deletion regions. Of the 61 known atypical NF1 deletions reported so far, 31 exhibit breakpoints which are located beyond one or both boundaries of the type-1 NF1 deletions. Of these 31 deletions, 27 are larger than 1.4-Mb and encompass more than the 14 protein-coding genes located within the type-1 NF1 deletion region (reviewed by Kehrer-Sawatzki et al. submitted for publication). Hence, these large deletions are likely to be of lesser importance in terms of genotype/phenotype correlations since the loss of additional genes located beyond the boundaries of the type-1 NF1 microdeletion region probably gives rise to an even more complex clinical phenotype and may be associated with additional clinical features not frequently observed in patients with type-1 NF1 deletions. In any case, these large atypical NF1 deletions can be detected by MLPA but neither the precise breakpoints nor the exact number of genes located within the deletion region can be ascertained using MLPA as the sole method of analysis. Owing to the limited number of MLPA-probes included in the SALSA® MLPA® Probemix P122-D2 NF1-area, the accurate characterization of these deletions in terms of their extent is not possible by MLPA, and only feasible by a method such as microarray analysis that is well suited to detect chromosomal aberrations at high resolution (Table 4).
In contrast to the 31 atypical NF1 deletions with breakpoints located beyond one or both boundaries of the type-1 NF1 deletions, a second group of atypical NF1 deletions has been identified which is characterized by breakpoints located within the type-1 NF1 microdeletion region (Pasmant et al. 2010; Vogt et al. 2012, 2014; Bianchessi et al. 2015; Zhang et al. 2015; Ferrari et al. 2017; Parisien-La Salle et al. 2019; Serra et al. 2019; Büki et al. 2021; Kehrer-Sawatzki et al. submitted for publication). These deletions are shorter than type-1 deletions and encompass only a subset of the 14 protein-coding genes located within the type-1 NF1 deletion region (Figs. 2 and 3). These deletions have been termed atypical group #2 deletions (Kehrer-Sawatzki et al. submitted for publication). So far, 30 atypical group #2 NF1 deletions have been identified (Figs. 2 and 3). By means of MLPA, these deletions can be distinguished from type-1, and in most instances also from type-3 deletions. Of these 30 atypical deletions, 21 can be also distinguished from type-2 deletions using MLPA as a single method of analysis. The relative extents of these 21 atypical deletions are indicated in Fig. 2. However, 9 of the 30 deletions cannot be distinguished from type-2 deletions by means of MLPA (Table 2). These 9 deletions represent a subgroup of group #2 deletions and are termed group #2A deletions. Their deletion boundaries are schematically indicated in Fig. 3. Breakpoint analysis revealed that the breakpoints of these 9 deletions are not located within SUZ12P and SUZ12 as would be characteristic of type-2 deletions (Vogt et al. 2012, 2014; Parisien-La Salle et al. 2019; Büki et al. 2021) (Table 3). Thus, atypical group #2A deletions and type-2 NF1 deletions cannot be distinguished from one another by MLPA. They do, however, differ from one another in terms of the genes they encompass. None of the 9 atypical group #2A deletions includes the SUZ12 gene. By contrast, the telomeric breakpoints of type-2 deletions are located within SUZ12 which is thereby functionally inactivated.

Schema of the type-1 NF1 microdeletion region, which includes 14 protein-coding genes as well as the SUZ12P pseudogene and 5 microRNA genes. The relative locations of these genes are indicated by black rectangles. Indicated below is the extent of the 21 known atypical group #2 NF1 deletions represented by vertical black bars. The patient IDs are indicated on the left. The low-copy repeats, NF1-REPa and NF1-REPc, are located at the boundaries of the type-1 NF1 microdeletion region. The atypical NF1 deletions of group #2, which exhibit breakpoints located within the boundaries of the type-1 NF1 microdeletion region, are smaller than type-1 NF1 deletions and do not encompass all of the genes located within the type-1 NF1 microdeletion region. As yet, 30 atypical group #2 NF1 deletions have been reported; indicated are 21 of these atypical group #2 deletions which can be distinguished from type-1 NF1 deletions by MLPA. Patients with IDs indicated in red were analysed by Vogt et al. (2012, 2014), those indicated in blue were analysed by Pasmant et al. (2010), in pink by Bianchessi et al. (2015) and in green by Zhang et al. (2015). Patient 171 was analysed by Ferrari et al. (2017), patients #1 and #2 by Serra et al. (2019), patients 134/260 by Büki et al. (2021) and patient 310221 by Kehrer-Sawatzki et al. submitted for publication). The deletions of the 5 patients whose IDs are marked by an asterisk do not encompass 4 of the 5 microRNA genes located within the type-1 NF1 microdeletion region. cen centromeric; tel telomeric

Schema of the type-1 NF1 microdeletion region, which includes 14 protein-coding genes as well as the SUZ12P pseudogene and 5 microRNA genes. The relative locations of these genes are indicated by black rectangles. Indicated below is the extent of the 9 known atypical group #2A NF1 deletions which are represented by vertical black bars. Patient IDs are indicated on the left. The low-copy repeats, NF1-REPa and NF1-REPc, are located at the boundaries of the type-1 NF1 microdeletion region. The atypical NF1 deletions of group #2A, which exhibit breakpoints located within the boundaries of the type-1 NF1 microdeletion region, cannot be distinguished from type-2 deletions using MLPA. The vertical red and green arrows represent the binding sites of the MLPA-probes included in the SALSA® MLPA® Probemix P122-D2 NF1-area (MRC-Holland). Red arrows represent MLPA-probes targeting genomic regions encompassed by the respective heterozygous deletions whereas green arrows represent MLPA-probes targeted to regions which are not deleted and present in two copies. The MLPA-probe SUZ12P intron 4 is shaded in green/red because the region targeted by this probe is deleted only in the patients marked by an asterisk, and not in the other 5 atypical group #2A deletions which are depicted here. cen: centromeric; tel: telomeric
Taken together, 9 (30%) of the 30 atypical deletions of group 2 cannot be accurately classified by MLPA. This is particularly relevant in view of the fact that the atypical group #2A NF1 deletions represent those deletions which are most important in the context of genotype/phenotype correlations since they may encompass only a subset of the 14 protein-coding and 5 microRNA genes located within the type-1 NF1 deletion interval.
Discussion
Patients with type-1 NF1 deletions often exhibit a severe clinical phenotype characterized by features that are not frequently seen in patients with intragenic pathogenic NF1 variants such as dysmorphic facial features, severe global developmental delay, cognitive disability, increased MPNST risk and a high number (as well as an accelerated growth rate) of neurofibromas (reviewed by Kehrer-Sawatzki et al. 2017, 2020; Ottenhoff et al. 2020; Büki et al. 2021; Pasmant et al. 2021; Pacot et al. 2021; Well et al. 2021). Genes located within the type-1 NF1 deletion interval and co-deleted with NF1 are likely to be responsible for the severe NF1 microdeletion-associated phenotype giving rise to the NF1 microdeletion syndrome. Importantly, the vast majority of NF1 microdeletion patients clinically characterized to date had a type-1 NF1 deletion, which is the most common type of NF1 deletion, observed in 70–80% of NF1 deletion patients.
Considerably less is known about the clinical phenotype in patients with other types of NF1 deletion. This may be due to the fact that these deletions occur much less frequently than type-1 NF1 deletions. Furthermore, type-2 NF1 deletions as well as atypical NF1 deletions are more frequently associated with somatic mosaicism with normal cells lacking the deletion; this may lead to a milder form of the disease. Somatic mosaicism is likely to be associated with highly variable clinical consequences depending not only on the NF1 microdeletion type, but also on the developmental stage when it has arisen, the cell types involved and the proportion of cells affected. This will certainly limit our ability to relate the clinical phenotype to the mutant genotype. To date, only 5 patients with non-mosaic type-2 NF1 deletions have been clinically characterized (Vogt et al. 2011; Zhang et al. 2015; Büki et al. 2021; Yethindra et al. 2021). From these data, it may be concluded that non-mosaic type-2 NF1 deletions are associated with a severe clinical phenotype similar to that exhibited by patients with type-1 NF1 deletions. However, further studies involving additional patients with non-mosaic type-2 deletions, accurately classified by methods with higher resolution than MLPA, would be necessary to confirm this.
By the same token, only a few patients with atypical NF1 deletions shorter than 1.4-Mb have been clinically characterized in any detail). The paucity of such patients has hampered the establishment of genotype/phenotype correlations. Patients with atypical NF1 deletions that do not encompass all of the genes located within the type-1 NF1 microdeletion interval may be very informative in terms of identifying potential modifier genes that could contribute to the severe phenotype observed in type-1 NF1 deletions. The clinical data available for 6 patients with such atypical group #2 deletions may provide support for this postulate since they manifested a less severe phenotype than that observed in patients with type-1 NF1 deletions (Kehrer-Sawatzki et al. submitted for publication).
To establish meaningful genotype/phenotype correlations, an accurate classification of NF1 microdeletions is crucial since the different types of NF1 deletion are associated with the loss of different numbers of flanking genes. The type-1 NF1 microdeletion region encompasses 14 protein-coding genes and 5 microRNA genes (miR-4733, miR-4724, miR-193A, miR-4725 and miR-365B). Whilst type-1, 2 and 3 deletions are invariably associated with the loss of all five microRNA genes, some atypical NF1 deletions do not include all five (Fig. 2). Hemizygosity for some of these microRNA genes may well be of clinical importance. The best characterized of the five microRNA genes within the NF1 gene region is miR-193A which is known to possess tumour suppressor functions (Jin et al. 2019; Chen et al. 2020; Polini et al. 2020; Wei et al. 2021). Hemizygosity for this microRNA gene may therefore facilitate tumour growth in those patients with NF1 microdeletions that encompass it. Five of the 30 atypical NF1 deletions of group #2 analysed to date do not include miR-193A (Fig. 2). However, the patients harbouring these deletions were either very young or clinically not characterized in any detail and hence it would be premature to discuss numbers of neurofibromas and their growth rates.
Five of the 14 protein-coding genes located within the type-1 NF1 deletion region, ATAD5, NF1, OMGP, RAB11FIP4 and SUZ12, are suspected to be loss-of-function intolerant since they exhibit ‘probability of loss-of-function” (pLI) scores of 0.99 or 1.0 (Supp. Table S1) (Lek et al. 2016). Consequently, hemizygosity for these genes is highly likely to exert a detrimental impact on the clinical phenotype in patients with deletions that encompass these genes. The different types of NF1 microdeletion are associated with variable copy numbers of these genes (Supp. Table S1). The SUZ12 gene, with a pLI score of 1.0, is very likely to be an important modifier of the type-1 NF1 microdeletion-associated phenotype. In patients with NF1 microdeletions, the loss of SUZ12 has been shown to increase the risk of MPNSTs (De Raedt et al. 2014; Lee et al. 2014; Zhang et al. 2014). Further, biallelic loss of SUZ12 is important in MPNST tumorigenesis (De Raedt et al. 2014). Patients with pathogenic variants located within SUZ12 but without NF1 microdeletions exhibit overgrowth, facial dysmorphic features, musculoskeletal abnormalities and developmental delay/intellectual disability (Imagawa et al. 2018; Cyrus et al. 2019a, b; Choufani et al. 2020). Thus, the loss of SUZ12 is likely to contribute significantly to the facial dysmorphic features, overgrowth, severe global developmental delay and the cognitive disabilities which are frequently noted in patients with type-1 NF1 deletions or other types of NF1 microdeletion that encompass the suppressor of zeste (SUZ12) gene. These clinical features are not observed in patients with intragenic pathogenic NF1 variants who possess two functional copies of the SUZ12 gene. Hence, the loss of SUZ12 in patients with NF1 microdeletions is most likely causally associated with these clinical features. This conclusion is supported by the clinical phenotype of patients with atypical group #2 deletions who are not hemizygous for a SUZ12 deletion (Kehrer-Sawatzki et al. submitted for publication). However, only 6 of these patients have been clinically characterized to date and further studies are necessary to corroborate this putative genotype/phenotype correlation.
A specific role in the development of the NF1 microdeletion-associated phenotype has recently been demonstrated for the cytokine receptor-like factor 3 (CRLF3) gene located in the centromeric part of the NF1 microdeletion region (Fig. 1). Induced pluripotent stem cell-forebrain cerebral organoids (hCOs), isolated from patients with type-1 NF1 microdeletions, display both neural stem cell proliferation and elevated neuronal abnormalities such as dendritic maturation deficits. Whilst increased neuronal stem cell proliferation has been shown to result from decreased NF1/RAS regulation, the neuronal differentiation, survival and maturation defects of these hCOs are caused by reduced CRLF3 expression and impaired RhoA signalling (Wegscheid et al. 2021). This role of CRLF3 has been corroborated by the observation that hCOs, isolated from a patient with an atypical NF1 deletion not encompassing the CRLF3 gene, did not exhibit abnormalities of neuronal survival, differentiation and maturation (Wegscheid et al. 2021). Further, these authors identified 7 of 17 NF1 patients with an increased autistic trait burden who harboured a germline missense putatively pathogenic variant within the CRLF3 gene (c.1166T>C, p.Leu389Pro) present in addition to pathogenic variants in the NF1 gene. Taken together, these findings indicate an essential role for CRLF3 in both human brain development and autism (Wegscheid et al. 2021). Indeed, a high autistic trait burden has been observed in children with type-1 NF1 deletions associated with the loss of one CRLF3 gene copy (Kehrer-Sawatzki et al. 2020). As yet, the clinical phenotype of patients with atypical NF1 deletions that do not encompass the CRLF3 gene has not been characterized in any detail with regard to the presence or absence of autistic traits. Further analyses of patients with deletions of this type will be necessary to determine the contribution of the CRLF3 gene to the NF1 microdeletion-associated phenotype.
The MLPA® Probemix P122-D2 NF1-area has turned out to be a valuable means to identify NF1 microdeletions and to characterize them, at least to a certain extent. A classification of NF1 deletions by MLPA is possible for type-1 deletions as shown by Summerer et al. (2018). According to their analysis, 99% of NF1 deletions initially identified by MLPA were indeed type-1 NF1 deletions as determined by sequence analysis of the breakpoints. By contrast, type-2 NF1 deletions and certain atypical NF1 deletions, those of group #2A, cannot be distinguished by MLPA; thus, further higher resolution analytical methods such as microarray analysis are required to determine the breakpoints and hence the number of genes included in the corresponding deletion intervals. The precise extent of NF1 deletions is critically important for establishing genotype/phenotype correlations and identifying potential modifier genes. Guidelines indicating the appropriate methods to use for the accurate classification of the different types of NF1 microdeletion are presented in Table 4. Correct classification of the deletion type is a prerequisite for being able to predict the potential presence of somatic mosaicism with normal cells. Type-1 NF1 deletions are only very rarely mosaic and the vast majority are germline deletions (Messiaen et al. 2011; Summerer et al. 2019). This is in accordance with the analysis of the parental origin of these deletions and their underlying mutational mechanism. It has been shown that 71% of type-1 NF1 microdeletions are caused by interchromosomal unequal crossover between either maternal or paternal chromosomes which confirms that these deletions are predominantly of meiotic origin. In most instances, type-1 NF1 deletions arise in the maternal germline (Lopez-Correa et al. 2000; Neuhäusler et al. 2018).
By contrast, somatic mosaicism is frequent in patients with type-2 and atypical NF1 deletions (Steinmann et al. 2007; Messiaen et al. 2011; Roehl et al. 2012; Vogt et al. 2014) (Table 4). Indeed, it has been estimated that at least 70% of all type-2 NF1 deletions are of postzygotic origin (Steinmann et al. 2007; Messiaen et al. 2011). So far, only 5 patients with non-mosaic type-2 NF1 deletions have been reported (Vogt et al. 2011; Zhang et al. 2015; Büki et al. 2021; Yethindra et al. 2021). The reason why the paralogous sequences SUZ12 and SUZ12P are more often involved in mitotic NAHR and only rarely in meiotic NAHR giving rise to non-mosaic type-2 NF1 deletions, are unclear. Genomic regions of increased NAHR activity giving rise to chromosomal aberrations have been reported to experience frequent allelic homologous recombination (AHR) during meiosis (Torres-Juan et al. 2007). Further, both the NAHR hotspots PRS1 and PRS2, located within the NF1-REPs, and those located within the CMT1A-REPs, overlap with pre-existing AHR hotspots (De Raedt et al. 2006; Lindsay et al. 2006). By contrast, the meiotic AHR activity within the region of SUZ12 and SUZ12P is low (Mussotter et al. 2014) which may explain the relatively rare occurrence of meiotic NAHR events causing non-mosaic type-2 NF1 deletions.
Mosaicism with normal cells has been shown to influence the clinical phenotype in patients with NF1 microdeletions, often leading to a mild manifestation of the disease (Kehrer-Sawatzki et al. 2012; Taylor Tovares et al. 2013). Hence the accurate classification of the type of NF1 microdeletion by methods such as microarray analysis may also help to identify patients with a high probability of being mosaic which might then require analysis of tissues other than blood in order to confirm or exclude mosaicism (Table 4). The assessment of mosaicism with normal cells without the deletion especially in founder patients carrying an NF1 microdeletion other than type 1 is very important with respect to deriving genotype/phenotype correlations. This assessment requires methodologies other than MLPA or microarray-analysis that are able to determine cells with and without the deletion at high resolution. Low-grade mosaicism with normal cells may be overlooked in patients with NF1 deletions investigated by MLPA as the single method of analysis since the intrinsic detection limit of mosaicism is in the range of 10–20% (reviewed by Summerer et al. 2019). If patients have high proportions of cells with the NF1 microdeletion in their blood, normal cells present at proportions lower than 10–20% are not going to be detectable by MLPA. Similar detection limits are associated with other methods such as microarray analysis and Sanger sequencing (reviewed by Summerer et al. 2019). Therefore, quantitative methods are necessary to detect or exclude low-grade mosaicism in patients with NF1 microdeletions, e.g. quantitative PCR (qPCR), droplet digital PCR (ddPCR), deep next-generation sequencing and fluorescence in situ hybridization (FISH) on a large number of cells. These methods, performed using different cell types such as blood lymphocytes and skin fibroblasts, may reliably establish whether the NF1 microdeletion is mosaic or not, which is important not only in relation to the observed phenotype in the patient, but also for the anticipated severity of the disease in the next generation, and the recurrence risk for siblings.
Conclusion
MLPA is a valuable method for the identification of large NF1 deletions but it has its limitations with regard to the accurate classification of the different NF1 microdeletion types. Whereas type-1 NF1 deletions appear to be classified relatively precisely by MLPA, this method fails to distinguish between type-2 and atypical group #2A NF1 deletions which represent 30% of all atypical NF1 deletions with breakpoints located within the type-1 NF1 microdeletion region (atypical NF1 deletions of group #2). Patients with atypical group #2 NF1 deletions may facilitate the establishment of genotype/phenotype correlations if they are associated with hemizygosity for only a subgroup of the genes located within the type-1 NF1 microdeletion interval. The precise characterization of these deletions in terms of their extent is a prerequisite for identifying such correlations.
Availability of data and material
The manuscript does not have associated data.
Code availability
The manuscript does not include software application or custom codes.
References
Bengesser K, Cooper DN, Steinmann K, Kluwe L, Chuzhanova NA, Wimmer K, Tatagiba M, Tinschert S, Mautner VF, Kehrer-Sawatzki H (2010) A novel third type of recurrent NF1 microdeletion mediated by nonallelic homologous recombination between LRRC37B-containing low-copy repeats in 17q11.2. Hum Mutat 31:742–751. https://doi.org/10.1002/humu.21254
Bengesser K, Vogt J, Mussotter T, Mautner VF, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2014) Analysis of crossover breakpoints yields new insights into the nature of the gene conversion events associated with large NF1 deletions mediated by nonallelic homologous recombination. Hum Mutat 35:215–226. https://doi.org/10.1002/humu.22473
Bianchessi D, Morosini S, Saletti V, Ibba MC, Natacci F, Esposito S, Cesaretti C, Riva D, Finocchiaro G, Eoli M (2015) 126 novel mutations in Italian patients with neurofibromatosis type 1. Mol Genet Genom Med 3:513–525. https://doi.org/10.1002/mgg3.161
Büki G, Zsigmond A, Czakó M, Szalai R, Antal G, Farkas V, Fekete G, Nagy D, Széll M, Tihanyi M, Melegh B, Hadzsiev K, Bene J (2021) Genotype-phenotype associations in patients With type-1, type-2, and atypical NF1 microdeletions. Front Genet 12:673025. https://doi.org/10.3389/fgene.2021.673025
Chen ZG, Zhao HJ, Lin L, Liu JB, Bai JZ, Wang GS (2020) Circular RNA CirCHIPK3 promotes cell proliferation and invasion of breast cancer by sponging miR-193a/HMGB1/PI3K/AKT axis. Thorac Cancer 11:2660–2671. https://doi.org/10.1111/1759-7714.13603
Choufani S, Gibson WT, Turinsky AL, Chung BHY, Wang T, Garg K, Vitriolo A, Cohen ASA, Cyrus S, Goodman S, Chater-Diehl E, Brzezinski J, Brudno M, Ming LH, White SM, Lynch SA, Clericuzio C, Temple IK, Flinter F, McConnell V, Cushing T, Bird LM, Splitt M, Kerr B, Scherer SW, Machado J, Imagawa E, Okamoto N, Matsumoto N, Testa G, Iascone M, Tenconi R, Caluseriu O, Mendoza-Londono R, Chitayat D, Cytrynbaum C, Tatton-Brown K, Weksberg R (2020) DNA methylation signature for EZH2 functionally classifies sequence variants in three PRC2 complex genes. Am J Hum Genet 106:596–610. https://doi.org/10.1016/j.ajhg.2020.03.008
Cnossen MH, van der Est MN, Breuning MH, van Asperen CJ, Breslau-Siderius EJ, van der Ploeg AT, de Goede-Bolder A, van den Ouweland AM, Halley DJ, Niermeijer MF (1997) Deletions spanning the neurofibromatosis type 1 gene: implications for genotype-phenotype correlations in neurofibromatosis type 1? Hum Mutat 9:458–464. https://doi.org/10.1002/(SICI)1098-1004(1997)9:5%3c458::AID-HUMU13%3e3.0.CO;2-1
Cyrus SS, Cohen ASA, Agbahovbe R, Avela K, Yeung KS, Chung BHY, Luk HM, Tkachenko N, Choufani S, Weksberg R, Lopez-Rangel E, C.A.U.S.E.S. Study, Brown K, Saenz MS, Svihovec S, McCandless SE, Bird LM, Garcia AG, Gambello MJ, McWalter K, Schnur RE, An J, Jones SJM, Bhalla SK, Pinz H, Braddock SR, Gibson WT (2019a) Rare SUZ12 variants commonly cause an overgrowth phenotype. Am J Med Genet C Semin Med Genet 181:532–547. https://doi.org/10.1002/ajmg.c.31748
Cyrus S, Burkardt D, Weaver DD, Gibson WT (2019b) PRC2-complex related dysfunction in overgrowth syndromes: a review of EZH2, EED, and SUZ12 and their syndromic phenotypes. Am J Med Genet C Semin Med Genet 181:519–531. https://doi.org/10.1002/ajmg.c.31754
De Luca A, Bottillo I, Dasdia MC, Morella A, Lanari V, Bernardini L, Divona L, Giustini S, Sinibaldi L, Novelli A, Torrente I, Schirinzi A, Dallapiccola B (2007) Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J Med Genet 44:800–808. https://doi.org/10.1136/jmg.2007.053785
De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F, Mautner V, Frahm S, Sciot R, Legius E (2003) Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 72:1288–1292. https://doi.org/10.1086/374821
De Raedt T, Stephens M, Heyns I, Brems H, Thijs D, Messiaen L, Stephens K, Lazaro C, Wimmer K, Kehrer-Sawatzki H, Vidaud D, Kluwe L, Marynen P, Legius E (2006) Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat Genet 38:1419–1423. https://doi.org/10.1038/ng1920
De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, Clapp W, Bradner J, Vidaud M, Upadhyaya M, Legius E, Cichowski K (2014) PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514:247–251. https://doi.org/10.1038/nature13561
Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K (2000) NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet 9:35–46. https://doi.org/10.1093/hmg/9.1.35
Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A (2002) Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 39:311–314. https://doi.org/10.1136/jmg.39.5.311
Evans DG, Huson SM, Birch JM (2012) Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res 2:17. https://doi.org/10.1186/2045-3329-2-17
Ferrari L, Scuvera G, Tucci A, Bianchessi D, Rusconi F, Menni F, Battaglioli E, Milani D, Riva P (2017) Identification of an atypical microdeletion generating the RNF135-SUZ12 chimeric gene and causing a position effect in an NF1 patient with overgrowth. Hum Genet 136:1329–1339. https://doi.org/10.1007/s00439-017-1832-5
Forbes SH, Dorschner MO, Le R, Stephens K (2004) Genomic context of paralogous recombination hotspots mediating recurrent NF1 region microdeletion. Genes Chromosomes Cancer 41:12–15. https://doi.org/10.1002/gcc.20065
Giannuzzi G, Siswara P, Malig M, Marques-Bonet T, NISC Comparative Sequencing Program, Mullikin JC, Ventura M, Eichler EE (2013) Evolutionary dynamism of the primate LRRC37 gene family. Genome Res 23:46–59. https://doi.org/10.1101/gr.138842.112
Hillmer M, Wagner D, Summerer A, Daiber M, Mautner VF, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2016) Fine mapping of meiotic NAHR-associated crossovers causing large NF1 deletions. Hum Mol Genet 25:484–496. https://doi.org/10.1093/hmg/ddv487
Hillmer M, Summerer A, Mautner VF, Högel J, Cooper DN, Kehrer-Sawatzki H (2017) Consideration of the haplotype diversity at nonallelic homologous recombination hotspots improves the precision of rearrangement breakpoint identification. Hum Mutat 38:1711–1722. https://doi.org/10.1002/humu.23319
Imagawa E, Albuquerque EVA, Isidor B, Mitsuhashi S, Mizuguchi T, Miyatake S, Takata A, Miyake N, Boguszewski MCS, Boguszewski CL, Lerario AM, Funari MA, Jorge AAL, Matsumoto N (2018) Novel SUZ12 mutations in Weaver-like syndrome. Clin Genet 94:461–466. https://doi.org/10.1111/cge.13415
Jenne DE, Tinschert S, Reimann H, Lasinger W, Thiel G, Hameister H, Kehrer-Sawatzki H (2001) Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am J Hum Genet 69:516–527. https://doi.org/10.1086/323043
Jin L, Li H, Wang J, Lin D, Yin K, Lin L, Lin Z, Lin G, Wang H, Ying X, Wang L, Zhang Y, Teng L (2019) MicroRNA-193a-5p exerts a tumor suppressor role in glioblastoma via modulating NOVA1. J Cell Biochem 120:6188–6197. https://doi.org/10.1002/jcb.27906
Kehrer-Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer U, Peyrl A, Jenne DE, Hansmann I, Mautner VF (2004) High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet 75:410–423. https://doi.org/10.1086/423624
Kehrer-Sawatzki H, Vogt J, Mußotter T, Kluwe L, Cooper DN, Mautner VF (2012) Dissecting the clinical phenotype associated with mosaic type-2 NF1 microdeletions. Neurogenetics 13:229–236. https://doi.org/10.1007/s10048-012-0332-y
Kehrer-Sawatzki H, Mautner VF, Cooper DN (2017) Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet 136:349–376. https://doi.org/10.1007/s00439-017-1766-y
Kehrer-Sawatzki H, Kluwe L, Salamon J, Well L, Farschtschi S, Rosenbaum T, Mautner VF (2020) Clinical characterization of children and adolescents with NF1 microdeletions. Childs Nerv Syst 36:2297–2310. https://doi.org/10.1007/s00381-020-04717-0
Kluwe L, Siebert R, Gesk S, Friedrich RE, Tinschert S, Kehrer-Sawatzki H, Mautner VF (2004) Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat 23:111–116. https://doi.org/10.1002/humu.10299
Lammert M, Friedman JM, Kluwe L, Mautner VF (2005) Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol 141:71–74. https://doi.org/10.1001/archderm.141.1.71
Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, Qin LX, Viale A, Singer S, Zheng D, Berger MF, Chen Y, Antonescu CR, Chi P (2014) PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 46:1227–1232. https://doi.org/10.1038/ng.3095
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation Consortium (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. https://doi.org/10.1038/nature19057
Lindsay SJ, Khajavi M, Lupski JR, Hurles ME (2006) A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet 79:890–902. https://doi.org/10.1086/508709
López-Correa C, Brems H, Lázaro C, Marynen P, Legius E (2000) Unequal meiotic crossover: a frequent cause of NF1 microdeletions. Am J Hum Genet 66:1969–1974. https://doi.org/10.1086/302920
López-Correa C, Dorschner M, Brems H, Lázaro C, Clementi M, Upadhyaya M, Dooijes D, Moog U, Kehrer-Sawatzki H, Rutkowski JL, Fryns JP, Marynen P, Stephens K, Legius E (2001) Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet 10:1387–1392. https://doi.org/10.1093/hmg/10.13.1387
Mautner VF, Kluwe L, Friedrich RE, Roehl AC, Bammert S, Högel J, Spöri H, Cooper DN, Kehrer-Sawatzki H (2010) Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J Med Genet 47:623–630. https://doi.org/10.1136/jmg.2009.075937
Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, Garcia-Linares C, Cooper DN, Lazaro C, Kehrer-Sawatzki H (2011) Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1). Hum Mutat 32:213–219. https://doi.org/10.1002/humu.21418
Mussotter T, Bengesser K, Högel J, Cooper DN, Kehrer-Sawatzki H (2014) Population-specific differences in gene conversion patterns between human SUZ12 and SUZ12P are indicative of the dynamic nature of interparalog gene conversion. Hum Genet 133:383–401. https://doi.org/10.1007/s00439-013-1410-4
Neuhäusler L, Summerer A, Cooper DN, Mautner VF, Kehrer-Sawatzki H (2018) Pronounced maternal parent-of-origin bias for type-1 NF1 microdeletions. Hum Genet 137:365–373. https://doi.org/10.1007/s00439-018-1888-x
Ottenhoff MJ, Rietman AB, Mous SE, Plasschaert E, Gawehns D, Brems H, Oostenbrink R, ENCORE-NF1 Team, van Minkelen R, Nellist M, Schorry E, Legius E, Moll HA, Elgersma Y (2020) Examination of the genetic factors underlying the cognitive variability associated with neurofibromatosis type 1. Genet Med 22:889–897. https://doi.org/10.1038/s41436-020-0752-2
Pacot L, Vidaud D, Sabbagh A, Laurendeau I, Briand-Suleau A, Coustier A, Maillard T, Barbance C, Morice-Picard F, Sigaudy S, Glazunova OO, Damaj L, Layet V, Quelin C, Gilbert-Dussardier B, Audic F, Dollfus H, Guerrot AM, Lespinasse J, Julia S, Vantyghem MC, Drouard M, Lackmy M, Leheup B, Alembik Y, Lemaire A, Nitschké P, Petit F, Dieux Coeslier A, Mutez E, Taieb A, Fradin M, Capri Y, Nasser H, Ruaud L, Dauriat B, Bourthoumieu S, Geneviève D, Audebert-Bellanger S, Nizon M, Stoeva R, Hickman G, Nicolas G, Mazereeuw-Hautier J, Jannic A, Ferkal S, Parfait B, Vidaud M, Network MOTNF, Wolkenstein P, Pasmant E (2021) Severe phenotype in patients with large deletions of NF1. Cancers 13:2963. https://doi.org/10.3390/cancers13122963
Parisien-La Salle S, Dumas N, Rondeau G, Latour M, Bourdeau I (2019) Isolated pheochromocytoma in a 73-year-old man with no clinical manifestations of type 1 neurofibromatosis carrying an unsuspected deletion of the entire NF1 gene. Front Endocrinol (lausanne) 10:546. https://doi.org/10.3389/fendo.2019.00546
Pasmant E, Sabbagh A, Masliah-Planchon J, Haddad V, Hamel MJ, Laurendeau I, Soulier J, Parfait B, Wolkenstein P, Bièche I, Vidaud M, Vidaud D (2009) Detection and characterization of NF1 microdeletions by custom high resolution array CGH. J Mol Diagn 11:524–529. https://doi.org/10.2353/jmoldx.2009.090064
Pasmant E, Sabbagh A, Spurlock G, Laurendeau I, Grillo E, Hamel MJ, Martin L, Barbarot S, Leheup B, Rodriguez D, Lacombe D, Dollfus H, Pasquier L, Isidor B, Ferkal S, Soulier J, Sanson M, Dieux-Coeslier A, Bièche I, Parfait B, Vidaud M, Wolkenstein P, Upadhyaya M, Vidaud D, members of the NF France Network (2010) NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat 31:E1506-1518. https://doi.org/10.1002/humu.21271
Pasmant E, Parfait B, Luscan A, Goussard P, Briand-Suleau A, Laurendeau I, Fouveaut C, Leroy C, Montadert A, Wolkenstein P, Vidaud M, Vidaud D (2015) Neurofibromatosis type 1 molecular diagnosis: what can NGS do for you when you have a large gene with loss of function mutations? Eur J Hum Genet 23:596–601. https://doi.org/10.1038/ejhg.2014.145
Polini B, Carpi S, Doccini S, Citi V, Martelli A, Feola S, Santorelli FM, Cerullo V, Romanini A, Nieri P (2020) Tumor suppressor role of hsa-miR-193a-3p and -5p in cutaneous melanoma. Int J Mol Sci 21:6183. https://doi.org/10.3390/ijms21176183
Rasmussen SA, Colman SD, Ho VT, Abernathy CR, Arn PH, Weiss L, Schwartz C, Saul RA, Wallace MR (1998) Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet 35:468–471. https://doi.org/10.1136/jmg.35.6.468
Roehl AC, Vogt J, Mussotter T, Zickler AN, Spöti H, Högel J, Chuzhanova NA, Wimmer K, Kluwe L, Mautner VF, Cooper DN, Kehrer-Sawatzki H (2010) Intrachromosomal mitotic nonallelic homologous recombination is the major molecular mechanism underlying type-2 NF1 deletions. Hum Mutat 31:1163–1173. https://doi.org/10.1002/humu.21340
Roehl AC, Mussotter T, Cooper DN, Kluwe L, Wimmer K, Högel J, Zetzmann M, Vogt J, Mautner VF, Kehrer-Sawatzki H (2012) Tissue-specific differences in the proportion of mosaic large NF1 deletions are suggestive of a selective growth advantage of hematopoietic del(+/-) stem cells. Hum Mutat 33:541–550. https://doi.org/10.1002/humu.22013
Santoro C, Riccio S, Palladino F, Aliberti F, Carotenuto M, Zanobio M, Peduto C, Nigro V, Perrotta S, Piluso G (2021) A novel MEIS2 mutation explains the complex phenotype in a boy with a typical NF1 microdeletion syndrome. Eur J Med Genet 64:104190. https://doi.org/10.1016/j.ejmg.2021.104190
Serra G, Antona V, Corsello G, Zara F, Piro E, Falsaperla R (2019) NF1 microdeletion syndrome: case report of two new patients. Ital J Pediatr 45:138. https://doi.org/10.1186/s13052-019-0718-7
Steinmann K, Cooper DN, Kluwe L, Chuzhanova NA, Senger C, Serra E, Lazaro C, Gilaberte M, Wimmer K, Mautner VF, Kehrer-Sawatzki H (2007) Type 2 NF1 deletions are highly unusual by virtue of the absence of nonallelic homologous recombination hotspots and an apparent preference for female mitotic recombination. Am J Hum Genet 81:1201–1220. https://doi.org/10.1086/522089
Summerer A, Mautner VF, Upadhyaya M, Claes KBM, Högel J, Cooper DN, Messiaen L, Kehrer-Sawatzki H (2018) Extreme clustering of type-1 NF1 deletion breakpoints co-locating with G-quadruplex forming sequences. Hum Genet 137:511–520. https://doi.org/10.1007/s00439-018-1904-1
Summerer A, Schäfer E, Mautner VF, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2019) Ultra-deep amplicon sequencing indicates absence of low-grade mosaicism with normal cells in patients with type-1 NF1 deletions. Hum Genet 138:73–81. https://doi.org/10.1007/s00439-018-1961-5
Taylor Tavares AL, Willatt L, Armstrong R, Simonic I, Park S-M (2013) Mosaic deletion of the NF1 gene in a patient with cognitive disability and dysmorphic features but without diagnostic features of NF1. Am J Med Genet Part A 161A:1185–1188. https://doi.org/10.1002/ajmg.a.35853
Torres-Juan L, Rosell J, Sánchez-de-la-Torre M, Fibla J, Heine-Suñer D (2007) Analysis of meiotic recombination in 22q11.2, a region that frequently undergoes deletions and duplications. BMC Med Genet 8:14. https://doi.org/10.1186/1471-2350-8-14
Uusitalo E, Rantanen M, Kallionpää RA, Pöyhönen M, Leppävirta J, Ylä-Outinen H, Riccardi VM, Pukkala E, Pitkäniemi J, Peltonen S, Peltonen J (2016) Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol 34:1978–1986. https://doi.org/10.1200/JCO.2015.65.3576
Vogt J, Nguyen R, Kluwe L, Schuhmann M, Roehl AC, Mußotter T, Cooper DN, Mautner VF, Kehrer-Sawatzki H (2011) Delineation of the clinical phenotype associated with non-mosaic type-2 NF1 deletions: two case reports. J Med Case Rep 5(1):577. https://doi.org/10.1186/1752-1947-5-577
Vogt J, Mussotter T, Bengesser K, Claes K, Högel J, Chuzhanova N, Fu C, van den Ende J, Mautner VF, Cooper DN, Messiaen L, Kehrer-Sawatzki H (2012) Identification of recurrent type-2 NF1 microdeletions reveals a mitotic nonallelic homologous recombination hotspot underlying a human genomic disorder. Hum Mutat 33:1599–1609. https://doi.org/10.1002/humu.22171
Vogt J, Bengesser K, Claes KB, Wimmer K, Mautner VF, van Minkelen R, Legius E, Brems H, Upadhyaya M, Högel J, Lazaro C, Rosenbaum T, Bammert S, Messiaen L, Cooper DN, Kehrer-Sawatzki H (2014) SVA retrotransposon insertion-associated deletion represents a novel mutational mechanism underlying large genomic copy number changes with non-recurrent breakpoints. Genome Biol 15:R80. https://doi.org/10.1186/gb-2014-15-6-r80
Wegscheid ML, Anastasaki C, Hartigan KA, Cobb OM, Papke JB, Traber JN, Morris SM, Gutmann DH (2021) Patient-derived iPSC-cerebral organoid modeling of the 17q11.2 microdeletion syndrome establishes CRLF3 as a critical regulator of neurogenesis. Cell Rep 36:109315. https://doi.org/10.1016/j.celrep.2021.109315
Wei KL, Chou JL, Chen YC, Low JT, Lin GL, Liu JL, Chang TS, Chen WM, Hsieh YY, Yan PS, Chuang YM, Lin JMJ, Wu SF, Chiang MK, Li C, Wu CS, Chan MWY (2021) Epigenetic silencing of STAT3-targeted miR-193a, by constitutive activation of JAK/STAT signaling, leads to tumor progression through overexpression of YWHAZ in gastric cancer. Front Oncol 11:575667. https://doi.org/10.3389/fonc.2021.575667
Well L, Döbel K, Kluwe L, Bannas P, Farschtschi S, Adam G, Mautner VF, Salamon J (2021) Genotype-phenotype correlation in neurofibromatosis type-1: NF1 whole gene deletions lead to high tumor-burden and increased tumor-growth. PLoS Genet 17:e1009517. https://doi.org/10.1371/journal.pgen.1009517
Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglböck H, Maertens O, Messiaen L (2006) Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer 45:265–276. https://doi.org/10.1002/gcc.20289
Yethindra V, Tagaev T, Mamytova E, Mainazarova E, Dzhumakova C, Namazbekova A (2021) A rare case of patient with neurofibromatosis type 1 in a genotype-phenotype correlation revealing a submicroscopic deletion on the long arm of chromosome 17. Clin Case Rep 9:2397–2399. https://doi.org/10.1002/ccr3.4047
Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, Gokaslan ZL, Riggins GJ, Wolinksy JP, Wood LD, Montgomery EA, Hruban RH, Kinzler KW, Papadopoulos N, Vogelstein B, Bettegowda C (2014) Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46:1170–1172. https://doi.org/10.1038/ng.3116
Zhang J, Tong H, Fu X, Zhang Y, Liu J, Cheng R, Liang J, Peng J, Sun Z, Liu H, Zhang F, Lu W, Li M, Yao Z (2015) Molecular characterization of NF1 and neurofibromatosis type 1 genotype-phenotype correlations in a Chinese population. Sci Rep 5:11291. https://doi.org/10.1038/srep11291
Zickler AM, Hampp S, Messiaen L, Bengesser K, Mussotter T, Roehl AC, Wimmer K, Mautner VF, Kluwe L, Upadhyaya M, Pasmant E, Chuzhanova N, Kestler HA, Högel J, Legius E, Claes K, Cooper DN, Kehrer-Sawatzki H (2012) Characterization of the nonallelic homologous recombination hotspot PRS3 associated with type-3 NF1 deletions. Hum Mutat 33:372–383. https://doi.org/10.1002/humu.21644
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
HKS collected the data; HKS and DNC wrote and edited the manuscript in equal parts.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kehrer-Sawatzki, H., Cooper, D.N. Classification of NF1 microdeletions and its importance for establishing genotype/phenotype correlations in patients with NF1 microdeletions. Hum Genet 140, 1635–1649 (2021). https://doi.org/10.1007/s00439-021-02363-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02363-3