
Abstract
About 5–10% of neurofibromatosis type 1 (NF1) patients exhibit large genomic germline deletions that remove the NF1 gene and its flanking regions. The most frequent NF1 large deletion is 1.4 Mb, resulting from homologous recombination between two low copy repeats. This “type-1” deletion is associated with a severe clinical phenotype in NF1 patients, with several phenotypic manifestations including learning disability, a much earlier development of cutaneous neurofibromas, an increased tumour risk, and cardiovascular malformations. NF1 adjacent co-deleted genes could act as modifier loci for the specific clinical manifestations observed in deleted NF1 patients. Furthermore, other genetic modifiers (such as CNVs) not located at the NF1 locus could also modulate the phenotype observed in patients with large deletions. In this study, we analysed 22 NF1 deletion patients by genome-wide array-CGH with the aim (1) to correlate deletion length to observed phenotypic features and their severity in NF1 deletion syndrome, and (2) to identify whether the deletion phenotype could also be modulated by copy number variations elsewhere in the genome. We then review the role of co-deleted genes in the 1.4 Mb interval of type-1 deletions, and their possible implication in the main clinical features observed in this high-risk group of NF1 patients.
Similar content being viewed by others

Introduction
Neurofibromatosis constitutes an autosomal inherited condition associated with tumour development of the nervous system. NF1 (neurofibromatosis type 1) forms the most common type of this disorder, with an estimated incidence of 1:3000 live births worldwide and is caused by heterozygous loss-of-function variants of the NF1 gene at 17q11.2. The commonest NF1-associated tumours are benign peripheral nerve tumours that may be cutaneous, subcutaneous, or plexiform neurofibromas.
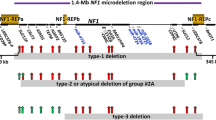
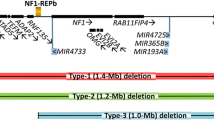
About 4–10% of NF1 patients carry large heterozygous genomic germline deletions that remove the NF1 tumour suppressor gene and its flanking regions [1]. Within this subsidiary, four delineated ‘types’ of NF1 deletion are observed and classified based on the length of deletion (Fig. 1) [2]. Type-1 deletions of 1.4 Mb are far more common than type-2 or type-3 deletions that span 1.2 Mb and 1.0 Mb respectively, whilst the other atypical deletions are more diverse [3]. Type-1 deletions mostly result from meiotic NAHR between NF1-REPa and NF1-REPc and encompass a 1.4 Mb region including 14 protein coding genes and 4 microRNA genes. Type-2 deletions are mediated by NAHR between SUZ12 and its pseudogene SUZ12P1, often occurring post-zygotically and leading to mosaicism [4, 5]. Type-3 deletions are less frequent (about 1–4% of all NF1 deletions) and are caused by NAHR between NF1-REPb and NF1-REPc [6, 7]. Non homologous end joining (NHEJ) or replication based mechanisms have been implicated in atypical deletions [3, 8,9,10,11].
Characterisation of the NF1 deletion syndrome is clinically of paramount importance due to the comparatively more severe clinical phenotype versus patients with NF1 point mutations [12]. A study of 29 patients [13] with type-1 NF1 deletions highlighted exacerbated features such as much earlier onset of cutaneous neurofibromas, dysmorphic facial features, large hands and feet, scoliosis, developmental delay, learning disabilities, accumulation of high tumour burden, cardiovascular malformations and a much higher risk of developing a malignant peripheral nerve sheath tumour (MPNST), an aggressive cancer associated with a poor prognosis [14]. MPNST lifetime risk across all NF1 patients is estimated to be 8–13%, but this estimation doubles to 16–26% in NF1 deletion patients [15]. Additionally, in direct contrast to the shortened stature frequently observed in patients exhibiting intragenic NF1 mutations, NF1 deletion patients more typically exhibit childhood overgrowth and tall stature in adults [16]. NF1 deletion patients significantly more often presented with symptomatic spinal neurofibromas, cardiovascular and skeletal abnormalities, learning disabilities, dysmorphism, and malignancies when compared to a “classic NF1” cohort, in a cohort of 126 NF1 deletions patients [1].
Undoubtedly, this severe clinical presentation in NF1 deletion patients could be correlated to the co-deleted genes in the region. Moreover, beyond considering these genes in isolation, another plausible mechanism could be that haploinsufficiency of these genes may synergize with each other, or with the NF1 loss, to induce the specific NF1 deletion syndrome (what may be called a contiguous gene syndrome). Additionally, whilst NF1 exhibits complete penetrance, its expressivity varies dramatically. Thus, establishing genotype to phenotype correlations could extract significant benefit in guiding pre-emptive clinical support and management of such an intractable disorder, particularly in the proportionally severe subset of NF1 deletion syndrome.
In this study, we analysed 22 NF1 deletion patients by genome-wide array-CGH with the aim (1) to correlate deletion length to observed phenotypic features and their severity in NF1 deletion syndrome, and (2) to identify whether the deletion phenotype could also be modulated by modifier copy number variations (CNVs) elsewhere in the genome. We also propose a review of the literature to suggest candidate modifier genes for the variable expressivity of NF1, among the genes located in the recurrent deletion interval.
Materials and methods
Patients and DNA analyses
We have analysed 22 unrelated NF1 deletion patients. DNA from these patients was tested using Human 1 M-Duo SNP chips (patients) and Human Omni1-Quad SNP chips (parents and sibling), respectively, according to manufacturer’s guidelines (Illumina, San Diego, CA). Briefly, for each sample ~ 200ng DNA were denatured, amplified, enzymatically fragmented, and hybridized to the BeadChips in a hybridization oven (Illumina) at 48˚C for 16-24 h. The BeadChips were washed according to the manufacturer’s protocol and the hybridized DNA subjected to primer extension with labelled nucleotides prior to detection using fluorescent antibodies. Data were processed using GenomeStudioV2009.2 (Illumina) and analysed using Nexus Discovery Edition v6.1 (BioDiscovery, Hawthorne, CA). All Nexus plots were inspected visually to verify calls made, identify uncalled events, and to exclude likely false positives. To exclude common germline CNVs, the Database of Genomic Variants (DGV), a comprehensive catalogue of structural variation in control data, was used. Copy number changes that encompassed changes noted in the DGV or identified in a clinically normal parent were excluded from further analysis. Regions of copy neutral loss of heterozygosity (cnLOH) were recorded only if > 5 Mb in size. This minimised the reporting of common cnLOH events that occurred in controls but would not rule out events arising through consanguinity. All genomic coordinates are given with reference to the GRCh36, hg18 assembly. Bioinformatic analysis of SNPs was also carried out. A correlation between SNPs and a specific clinical feature was explored.
Statistical analysis
Assessment was undertaken through the tabulation of summary statistics for values under consideration. Median (range) and frequencies (percentages) for the association between two covariates were used for the description of the relationship. The significance level for all statistical tests was 0.05. Note that the primary aim to identify modifying loci that interact with partial NF1-loss to induce a more severe phenotype in NF1 deletion syndrome uses multiple comparisons, and multiplicity problems could appear in this analysis. Multiplicity may inflate the type I error (α) and the probability of finding a significant association just by chance, a false-positive conclusion. Each test has a 5% chance of a false positive result when there is no real association (a type I error) so if the analysis has multiple comparisons the probability of at least one false positive result is very much greater than 5%. Consequently, the significance level for all statistical tests could be adjusted. A simple and intuitive multiplicity adjustment is the Bonferroni method, which requires that the p-value for each comparison be less than or equal to 0.05 divided by the total number of comparisons. In this analysis, the primary aim performs 112 comparisons, then the Bonferroni adjustment method would require a p-value less than or equal to 0.00045. The disadvantage of multiplicity adjustment methods is that they can be quite conservative if there are many comparisons and could increase β thereby reducing statistical power. The analysis was performed using the statistical software STATA11.
Results
Clinical data
We have analysed 22 unrelated NF1 deletion patients, including 18 patients with a type-1 deletion, three patients with a type-2 deletion, and one patient with an atypical > 5.5 Mb deletion. Clinical details were also recorded from these 22 patients.
Available clinical information for the 22 patients and comparison with the general NF1 population are summarised in Table 1; however, it should be noted that not all clinical information was available.
Analysis of five nuclear families with NF1 deletion child and unaffected parents failed to reveal additional novel CNVs in the proband. We then looked for CNVs in the combined 22 deletion patients. No significant findings were observed.
Association between NF1 deletion and patient features
Using NF1 length as continuous variable to assess its relationship with patient features, patients with learning difficulties were more likely to show higher NF1 deletion length at significant level p = 0.025 (Table 2, Mann-Whitney test). However, the number of patients was too small to draw a reliable conclusion. The length was not associated with any of other observed features.
‘Patient 2’ was not included in the analysis, as he exhibited an atypical deletion > 5.5 Mb far larger than the deletions observed in 18 patients with a type-1 deletion and 3 patients with a type-2 deletion.
Comparison of total NF1 deletion size > 1.2 Mb between each patient feature using Fisher’s exact test concluded that patients with learning difficulties showed higher percentage of NF1 deletion size > 1.2 Mb (100%) than patients without (0%) (p = 0.007, Fisher’s exact test; not significant after Bonferroni correction). Significance was not shown in any other features (Supplementary Tables S1 and S2). Same comparisons were carried out for total NF1 deletion size > 1.35 Mb between each patient feature, the results did not show any difference between patients with and without NF1 deletion size > 1.35 Mb, in relationship to the patient features (Supplementary Tables S3 and S4). In addition, there was no association shown in the comparisons for total NF1 deletion size > 1.40 Mb between each patient features (Supplementary Tables S5 and S6).
Association between gene loss in NF1 deletion and patient features
To assess the association between gene loss in NF1 deletion and patient features, results concluded that patients with learning difficulties showed higher number of genes loss in NF1 deletion than patients without learning difficulties at p-value equal to 0.0087 (Mann-Whitney test; not significant after Bonferroni correction) (Table 3). Please also note that the number in the ‘learning difficulties’ symptom is extremely small.
Association between genes loss in NF1 and other sites and patient features
Comparison of number of genes loss in NF1 deletion, other non-DGV, non-inherited, and other gains between each patient feature were carried out using non-parametric Mann-Whitney test. See Tables 4, 5 and 6 for details. Results concluded that patients with learning difficulties showed higher number of genes loss in NF1 region and other sites (statistically significant association, p-value < 0.05; not significant after Bonferroni correction). However, the number of patients without learning difficulties was, again, too small (n = 2) to draw a reliable conclusion. A borderline significance was observed between the loss of several genes and plexiform neurofibromas (Table 5), when Patient 2 excluded. This observation must be confirmed by a larger independent study.
Candidate modifier genes localized in the 1.4 mb type-1 deletion region and their putative role in clinical expressivity of NF1
The 1.4 Mb locus encompassed by NF1 type-1 deletions comprises 13 protein coding genes, and 5 microRNA genes. We summarize here what is known about these genes, and how they might be implicated in the severe phenotype evidenced in this group of patients (Table 7).
SUZ12
SUZ12 is located approximately 40 kb telomeric to the NF1 gene. Both type-1 and type-3 deletions encompass the gene, and recurrent recombination events with its pseudogene, SUZ12P1, is responsible for type-2 deletions (Fig. 1). Fusion transcript analysis in type-2 deletion patients identified chimeric sequences predicted to contain premature stop codons, presumably coding for truncated proteins, or no protein at all [18].
SUZ12 is a 739 amino-acid protein with ubiquitous expression. As a core subunit of the Polycomb Repressive Complex 2 (PRC2) together with embryonic ectoderm development (EED), the histone methyltransferase enhancer of zeste homologue 1/2 (EZH1/2), and with RB binding protein 4 or 7 (RBBP4 or RBBP7), the PRC2 catalyses the mono-, di- and tri-methylation of histone H3 at lysine 27, implicated in the maintenance of transcriptional repression of several target genes in a cell-type and differentiation-stage specific manner [19]. In vivo knock-out models have shown that PRC2 core proteins (EED, EZH1/EZH2, and SUZ12) are essential for embryonic development, as illustrated by embryonic lethality in Suz12−/− mouse around gastrulation [20].
In human, heterozygous loss-of-function variants affecting PRC2 core proteins genes are responsible for overgrowth with intellectual disability disorders (OGID) [21]. Weaver syndrome (WS) is caused by mutations in EZH2 (Enhancer of Zeste homolog 2) [22] and is characterized by pre- and/or postnatal overgrowth, macrocephaly, advanced bone age, distinctive craniofacial features, and a variable degree of intellectual disability. Musculoskeletal abnormalities are also frequently observed. Some patients develop childhood tumours [23,24,25]. Cohen-Gibson syndrome (COGIS) was described later, involving mutations in EED (Embryonic Ectoderm Development) [26], and can be distinguished from Weaver syndrome by more prevalent cryptorchidism, cervical spine abnormalities, and cardiac abnormalities [27]. The first case report for an heterozygous SUZ12 pathogenic variant in human was described in 2017 in an 11-year-old girl [28]. The variant was inherited from her father, who showed mosaicism for the mutation and had a milder clinical presentation. Later reports by the same group led the condition to become Imagawa-Matsumoto syndrome (IMMAS) [29]. Affected individuals develop malformations and disabilities generally milder than those observed in other OGID due to PRC2 mutations, with the exception of a more prominent increase in postnatal head circumference, and the presence in some but not all IMMAS patients of hypertrichosis, a condition never described in WS and COGIS [29]. A recently published patient with a 1.4 Mb deletion encompassing SUZ12, but not NF1, confirms the consequences of SUZ12 haploinsufficiency: large hands and feet, hyperlaxity, intellectual disability, macrocephaly, dysmorphism, and postnatal overgrowth [30]. Altogether, SUZ12 haploinsufficiency can more certainly be at least in part responsible for the learning disabilities and the overgrowth phenotype observed in some NF1-deleted patients.
Several studies have shown the role of PRC2 in central nervous system (CNS) development. In vitro or in vivo inactivation studies of either EZH1, EZH2, EED, or SUZ12 suggest their implication in neuronal maturation and migration, spinal cord development, synaptic plasticity, astrocyte or oligodendrocyte differentiation, and myelination of the central and peripheral nervous systems (reviewed by Liu et al. [31]).. As neurological issues have also been observed in Weaver and other overgrowth syndromes [21], this argues in favour of the implication of SUZ12 in learning disabilities in NF1 deletion patients.
Although constitutive depletion of PRC2 core proteins is responsible for embryonic lethality in mice, complete genetic loss- or gain-of-function variants in somatic tissues constitute a driver event for several tumour types through a major alteration of transcription regulation and the alteration of RAS, WNT, and NOTCH signalling [32], and can be at the origin of lymphoid and myeloid malignancies [33], as well as MPNST [34], this later being significantly more prevalent in NF1-deleted patients [15]. Hence, mutually exclusive bi-allelic loss-of-function mutations in the PRC2 core proteins SUZ12 and EED are recurrently observed in NF1-associated MPNSTs, while not observed in the pre-cancerous tumours, neurofibromas [35, 36]. It is then reasonable to postulate that constitutive heterozygous deletion of SUZ12 in NF1 deletion patient represents a risk factor for MPNST development in NF1 deletion patients.
PRC2 time- and tissue-specific epigenetic programming plays a major role in cell fate and organogenesis. Cardiac cell lineage inactivation of PRC2 components causes major cardiac malformations, eventually leading to neonatal lethality (see Wang et al. for review [37]). Comparable phenomenon is observed by selective inactivation of long noncoding RNAs interacting with PRC2, leading to altered cardiogenic gene transcription: Braveheart (Bvht) [38], Fetal-lethal noncoding developmental regulatory RNA (Fendrr) [39], CARdiac Mesoderm Enhancer-associated Noncoding RNA (Carmn) [40], cardiac-hypertrophy-associated epigenetic regulator (Chaer) [41], Ppp1r1b [42], human-specific heart brake lncRNA 1 (HBL1) [43]. The field is still vast and unexplored, and there is much to be done to understand the role of SUZ12 in the increased risk for cardiovascular malformations in NF1-deleted patients.
ATAD5
Constitutional mismatch repair (MMR) deficiency (CMMRD) is a tumour predisposition syndrome that shares some clinical features with NF1, and both syndromes are associated with the occurrence of café-au-lait spots, but also high-grade glioma, acute myeloid leukaemia or rhabdomyosarcoma [44]. ATPase family AAA domain-containing protein 5 (ATAD5) belongs to the proliferation cell nuclear antigen (PCNA) RFC-like (RLC) unloader complex, whose major role is to prevent accumulation of PCNA on chromatin after DNA synthesis [45, 46]. PCNA loading/unloading cycling is essential for proper cell cycle timing and genomic stability. It is presumably implicated in several DNA alterations and repair mechanisms, including double-strand breaks (DSB) repair, gross chromosomal rearrangements, and DNA damage tolerance (DDT) pathway, as well as in sister chromatid cohesion, and chromatin and telomere length maintenance.
PCNA is necessary for the recruitment of MMR proteins to the replication fork. In an in vitro model, inactivation of elg1, the yeast homolog for ATAD5, led to PCNA over-retention on DNA, and subsequent mutation accumulation mediated by improper recruitment of Msh2-Msh6 and Msh2-Msh3 heterodimers [47].
So far, heterozygous mutations in ATAD5 have been evidenced in endometrial, breast and ovarian cancers [48, 49]. Type-1 and type-2 deletions at the NF1 locus constitutively result in ATAD5 haploinsufficiency (Fig. 1), which most probably constitutes an additional risk factor for genomic instability and tumour emergence.
ADAP2
Originally named centaurin-α2 (CENTA2) for its amino acid identity with centaurin-α1, ADAP2 (ARFGAP with Dual Pleckstrin homology domains 2) is present in a wide variety of tissues. It appears to predominantly reside in the cytosol but can have a sustained localisation at the plasma membrane under activation of the PI 3-kinase. It functions as a GTPase activating protein (GAP) for ARF6 (ADP-ribosylation factor 6), a protein implicated in actin cytoskeleton remodelling [50]. ADAP2 is able to bind to microtubules and was suggested to act as microtubule-associated protein (MAP) increasing microtubule stability [51].
ADAP2 is expressed during key stages of heart formation during embryogenesis. Using a morpholino experiment in zebrafish, Venturin et al. [52]. showed that adap2 inactivation led to inappropriate heart jogging and heart looping, abnormal ventricular size and atrioventricular valve formation at 2 or 3 days post fertilisation. Incomplete alteration of splicing with an additional morpholino did not cause significant defects, suggesting that complete loss of adap2 would be required for cardiovascular malformations to occur. ADAP2 might thus play a role in the higher prevalence of cardiovascular malformations in NF1 deletion patients.
Also, ADAP2 is able to regulate type 1 interferon (IFN1) response during viral infection [53], a signalling pathway that might contribute to neurofibroma formation [54].
CRLF3
A publication in 2021 identified Cytokine receptor-like factor 3 (CRLF3) as a candidate gene for autistic traits in NF1 deleted patients [55]. They showed both a hyperproliferation of neural crest cells and an abnormal dendritic maturation in iPSC-cerebral organoid models derived from type-1 deleted cells. Neurogenic defects were also observed in CRLF3-inactivated organoids, pointing out to a specific role of the gene in the proper maturation of neurons, though the precise mechanism underlying such regulation remains to be elucidated.
A more recent study pointed out Crlf3 as a negative regulator of IFN1 and IFN-stimulated genes (ISG) production in Miichthys miiuy fish [56]. In their experiments, Crlf3 interacted with TANK-binding kinase 1 (TBK1) and promoted its degradation via ubiquitination, presumably preventing cell response to viral infection.
RNF135
Douglas et al. [57] identified in 2007 a truncating mutation or complete deletion of RNF135 in 5 individuals out of 245 unrelated individuals with overgrowth and learning disabilities, plus one patient with a missense variant in RNF135. Though this result was suggestive of a direct implication of RNF135 in overgrowth pathogenesis, Visser et al. [58]. did not identify any pathogenic variant in the gene in a cohort of 160 unrelated individuals with Sotos syndrome, but rather missense, synonymous or intronic variants of unknown significance. In a French cohort of patients with autism, Tastet et al. identified the same type of RNF135 variants, among which p.Arg115Lys appeared significantly over-represented in their cohort compared to control populations [59]. Still, this variant is reported as homozygous in 22 individuals in the gnomAD v2.1.1 database. The RNF135 gene encodes an E3 ubiquitin ligase, a pathway previously implicated in autism and intellectual disability. Available data leave the question of whether RNF135 is indeed responsible for overgrowth and learning difficulties open.
Overexpression of RNF135 inhibited the in vitro proliferation and invasiveness of SCC25 cells, a model of tongue carcinoma, suggesting a putative role in cancer regulation [60].
Other genes of the type-1 deletion interval
Over-expression of Oligodendrocyte-myelin glycoprotein (OMpg) resulted in inhibition of NSC proliferation in a model of neurospheres derived from mesencephalon of rat embryos [61], suggesting a complementarity with the role of neurofibromin in dendritic spine morphology and plasticity [62].
UTP6 Small Subunit Processome Component (UTP6), or Hepatocellular carcinoma antigen 66 (HCA66), is a positive regulator of Apaf-1-dependent apoptosis [63]. As such, its loss represents an additional mechanism by which tumour cells might escape from death in NF1 deletion patients.
There is still little known about the COPRS gene, whose interactions with protein-arginine methyltransferase 5 (PRTM5) and histone H4, and variable expressivity among MPNST cell lines, render its implication in tumoral process uncertain [64].
A few micro RNAs genes are present in the NF1 type-1 deletion interval: MIR4733, MIR4724, MIR193A, MIR4725, and MIR365B. miR-193a has well-known tumour suppressor functions, in hepatocellular carcinoma [65], endometrioid endometrial adenocarcinoma [66], non-small-cell lung cancer [67], and breast cancer [68]. Though poorly described in MPNST, miRNAs dysregulation holds a great potential for tumour progression and treatment.
Discussion
Despite exhibiting complete penetrance, phenotypic manifestations of NF1 may vary substantially. In NF1 deletion syndrome, the sources of this variation have not been confidently established, although the deletion itself has been proposed to contribute. The deletion length and by extension, the deleted adjacent genes may particularly contribute to the relative severity of NF1 deletion syndrome versus NF1 caused by intragenic mutations. In addition, due to the deletion mechanism, a subset of patients - particularly type-2 deletion [8] - exhibit mosaicism which itself intrinsically contributes to phenotypic variation.
The primary investigation of this study found no significant novel CNVs within the genomes of the 22 NF1 deletion patients. It was the first study of its type attempting to explore the presence of modifier CNVs in NF1 deletion patients. A preferable approach to investigating the existence of modifying loci would have been to examine gene expression for a functional analysis; but we were unable to source and experiment with RNA from these patient samples and therefore had no choice but to carry out an exploratory study into the sourced DNA samples.
A positive correlation was however noted within this study, though not significant after correction for multiple testing, between NF1 deletion length and learning disability, a clinical manifestation more prevalent in NF1 deletion patients [1, 71]; the power of this study is however extremely low and must therefore be treated with caution. All type-1 deletion patients for which neurodevelopmental data was available in this study (n = 16) exhibited learning disability; whilst all type-2 deletion patients for which neurodevelopmental data was available (n = 2) did not exhibit learning disability. Whilst conclusions are difficult to propose with such few patient numbers, a much lower incidence of learning disability could occur in type-2 deletion in comparison to type-1. Similarly, another study by Vogt et al. [72]. also investigated two type-2 deletion patients; neither of which exhibited neurodevelopmental retardation. Other studies with larger numbers of patient did not analyse this comparison. Pasmant et al. [14]. reported a 86% incidence of learning disability in type-1 deletion (n = 44) which was higher than the 80% incidence in all NF1 deletion (n = 58).
Establishing such a genotype-phenotype correlation could provide significant clinical benefit, especially in the frame of neurodevelopmental. Indeed, as discussed by Lemay et al. [73], encouraging rapid diagnosis aids in earlier provision of management, advice and support crucial to the patient and their families. This support may take the form of genetic counselling or referral to appropriate services for therapy, education, financial services, respite care, or early intervention support programmes. Indeed, respite care has been found to reduce the incidence of child maltreatment, whilst early intervention programmes before and during school years develop the patient’s language, behavioural, physical and self-management skills, and prepare the child for school [74]. Future study analysing the relative phenotypic incidence between deletion types may therefore provide clinically salient conclusions, perhaps contributing to screening procedures that inform subsequent management.
This potentially increased predisposition of type-1 deletion over type-2 in exhibiting the phenotype of learning disability may find its mechanistic basis in loss of genes such as LRRC37B located within the extra 0.2 Mb deleted in type-1 deletion (Fig. 1). It was also proposed that the recognisably less severe clinical phenotype in patients with type-2 NF1 deletion does not relate to the extent of the deletion but rather may be associated with the frequently observed mosaicism stemming from mitotic NAHR causing type-2 deletion, leading to the presence of normal cells that lack the deletion. Unless mosaicism can be quantified (in the relevant tissues), its presence further complicates translation of genotype-phenotype correlations to the clinical setting.
Several genes included in the type-1 deletion region have been suggested to account for the more severe phenotype observed in this group of patients (Table 7). They are predicted to contribute to cardiovascular malformations (SUZ12, ADAP2), higher malignant potential (UTP6, ATAD5, SUZ12, RNF135, COPRS, MIR193A, MIR365B), overgrowth (RNF135, SUZ12), intellectual disabilities (OMG, RNF135, SUZ12, CRLF3). Altogether, SUZ12, which is patently altered in the three main types of NF1 locus deletions, has emerged as the major candidate for most symptoms over-represented in NF1-deleted patients.
Overall, the major drawbacks of this study are the incomplete clinical data, inability to acquire sample RNA to directly assess gene expression and the low power. Another previously conducted study by Riva and collaborators [75] showed no significant association of any phenotype with deletion length; although this study was limited to even less patients (n = 10).
Future studies based on exome/whole genome re-sequencing may reveal modifying loci or signalling pathways leading to pharmaceutical targets, begetting further development of management. Further research also based on many rigorously clinically characterised deletion and non-deletion patients may highlight specific SNPs largely associated with a single clinical conglomeration of clinical features. Another important issue will be the phenotypic description. The impact of deleted genes does not fully explain the phenotype, and confounding biases involving other factors modulating the phenotype complicate genotype-phenotype correlations.
In addition, it is important to accurately report the phenotype and differentiate between clinical features, consequences, and complications, as emphasized by Vincent M Riccardi [76]. By distinguishing between different levels of phenotypic description, we can gain a better understanding of the causal genetic factors.
In conclusion, this study did not find any unique CNVs (copy number variations) in NF1 deletion syndrome. However, the study did show a positive correlation between the length of NF1 locus deletion and learning disabilities. These findings will help guide future research into establishing correlations between genotypes and phenotypes.

Illustration of the genomic region harboring the NF1 and adjacent genes. Red, green, and blue intervals represent the deletion extent in the recurrently observed type-1, type-2, and type-3 NF1 deletions respectively. NF1 is depicted in orange; SUZ12 and its pseudogene SUZ12P1 in green. Purple intervals indicate low-copy repeats (LCRs) implicated in non-allelic homologous recombination (NAHR) at the origin of type-1 and type-3 deletions. Arrows adjacent to gene symbols denote transcriptional orientation. Cen and tel refer to centromeric and telomeric direction, respectively
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Pacot L, Vidaud D, Sabbagh A, Laurendeau I, Briand-Suleau A, Coustier A, et al. Severe phenotype in patients with large deletions of NF1. Cancers (Basel). 2021;13:2963.
Kehrer-Sawatzki H, Cooper DN. Classification of NF1 microdeletions and its importance for establishing genotype/phenotype correlations in patients with NF1 microdeletions. Hum Genet. 2021;140:1635–49.
Vogt J, Bengesser K, Claes KBM, Wimmer K, Mautner V-F, van Minkelen R, et al. SVA retrotransposon insertion-associated deletion represents a novel mutational mechanism underlying large genomic copy number changes with non-recurrent breakpoints. Genome Biol. 2014;15:R80.
Bengesser K, Vogt J, Mussotter T, Mautner V-F, Messiaen L, Cooper DN, et al. Analysis of crossover breakpoints yields new insights into the nature of the gene conversion events associated with large NF1 deletions mediated by nonallelic homologous recombination. Hum Mutat. 2014;35:215–26.
Roehl AC, Vogt J, Mussotter T, Zickler AN, Spöti H, Högel J, et al. Intrachromosomal mitotic nonallelic homologous recombination is the major molecular mechanism underlying type-2 NF1 deletions. Hum Mutat. 2010;31:1163–73.
Zickler AM, Hampp S, Messiaen L, Bengesser K, Mussotter T, Roehl AC, et al. Characterization of the nonallelic homologous recombination hotspot PRS3 associated with type-3 NF1 deletions. Hum Mutat. 2012;33:372–83.
Pasmant E, Sabbagh A, Masliah-Planchon J, Haddad V, Hamel M-J, Laurendeau I, et al. Detection and characterization of NF1 microdeletions by custom high resolution array CGH. J Mol Diagn. 2009;11:524–9.
Vogt J, Mussotter T, Bengesser K, Claes K, Högel J, Chuzhanova N, et al. Identification of recurrent type-2 NF1 microdeletions reveals a mitotic nonallelic homologous recombination hotspot underlying a human genomic disorder. Hum Mutat. 2012;33:1599–609.
Pasmant E, de Saint-Trivier A, Laurendeau I, Dieux-Coeslier A, Parfait B, Vidaud M, et al. Characterization of a 7.6-Mb germline deletion encompassing the NF1 locus and about a hundred genes in an NF1 contiguous gene syndrome patient. Eur J Hum Genet. 2008;16:1459–66.
Pacot L, Pelletier V, Chansavang A, Briand-Suleau A, Burin des Roziers C, Coustier A, et al. Contribution of whole genome sequencing in the molecular diagnosis of mosaic partial deletion of the NF1 gene in neurofibromatosis type 1. Hum Genet. 2022. https://doi.org/10.1007/s00439-022-02476-3.
Kehrer-Sawatzki H, Wahlländer U, Cooper DN, Mautner V-F. Atypical NF1 microdeletions: challenges and opportunities for Genotype/Phenotype correlations in patients with large NF1 deletions. Genes (Basel). 2021;12:1639.
Bettegowda C, Upadhayaya M, Evans DG, Kim A, Mathios D, Hanemann CO, et al. Genotype-phenotype correlations in neurofibromatosis and their potential clinical use. Neurology. 2021;97(7 Suppl 1):91–8.
Mautner V-F, Kluwe L, Friedrich RE, Roehl AC, Bammert S, Högel J, et al. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 mb type-1 NF1 deletions. J Med Genet. 2010;47:623–30.
Pasmant E, Sabbagh A, Spurlock G, Laurendeau I, Grillo E, Hamel M-J, et al. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat. 2010;31:E1506–1518.
De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72:1288–92.
van Asperen CJ, Overweg-Plandsoen WC, Cnossen MH, van Tijn DA, Hennekam RC. Familial neurofibromatosis type 1 associated with an overgrowth syndrome resembling Weaver syndrome. J Med Genet. 1998;35:323–7.
Koczkowska M, Callens T, Chen Y, Gomes A, Hicks AD, Sharp A, et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: genotype-phenotype study in neurofibromatosis type 1. Hum Mutat. 2020;41:299–315.
Steinmann K, Cooper DN, Kluwe L, Chuzhanova NA, Senger C, Serra E, et al. Type 2 NF1 deletions are highly unusual by virtue of the absence of nonallelic homologous recombination hotspots and an apparent preference for female mitotic recombination. Am J Hum Genet. 2007;81:1201–20.
Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9.
Piunti A, Shilatifard A. The roles of polycomb repressive complexes in mammalian development and cancer. Nat Rev Mol Cell Biol. 2021;22:326–45.
Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, et al. Mutations in epigenetic regulation genes are a Major cause of overgrowth with intellectual disability. Am J Hum Genet. 2017;100:725–36.
Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, et al. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012;90:110–8.
Coulter D, Powell CM, Gold S. Weaver syndrome and neuroblastoma. J Pediatr Hematol Oncol. 2008;30:758–60.
Basel-Vanagaite L. Acute lymphoblastic leukemia in Weaver syndrome. Am J Med Genet A. 2010;152A:383–6.
Kelly TE, Alford BA, Abel M. Cervical spine anomalies and tumors in Weaver syndrome. Am J Med Genet. 2000;95:492–5.
Cohen ASA, Tuysuz B, Shen Y, Bhalla SK, Jones SJM, Gibson WT. A novel mutation in EED associated with overgrowth. J Hum Genet. 2015;60:339–42.
Griffiths S, Loveday C, Zachariou A, Behan L-A, Chandler K, Cole T, et al. EED and EZH2 constitutive variants: a study to expand the Cohen-Gibson syndrome phenotype and contrast it with Weaver syndrome. Am J Med Genet A. 2019;179:588–94.
Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, et al. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat. 2017;38:637–48.
Imagawa E, Seyama R, Aoi H, Uchiyama Y, Marcarini BG, Furquim I, et al. Imagawa-Matsumoto syndrome: SUZ12-related overgrowth disorder. Clin Genet. 2023. https://doi.org/10.1111/cge.14296.
Park S, Jang M-A. Identification of SUZ12 haploinsufficiency due to a 1.4-Mb deletion at 17q11.2 in a child with overgrowth and intellectual disability syndrome. Ann Lab Med. 2023;43:319–22.
Liu P-P, Xu Y-J, Teng Z-Q, Liu C-M. Polycomb repressive complex 2: emerging roles in the Central Nervous System. Neuroscientist. 2018;24:208–20.
Suppiah S, Mansouri S, Mamatjan Y, Liu JC, Bhunia MM, Patil V, et al. Multiplatform molecular profiling uncovers two subgroups of malignant peripheral nerve sheath tumors with distinct therapeutic vulnerabilities. Nat Commun. 2023;14:2696.
Li B, Chng W-J. EZH2 abnormalities in lymphoid malignancies: underlying mechanisms and therapeutic implications. J Hematol Oncol. 2019;12:118.
Wassef M, Margueron R. The multiple facets of PRC2 alterations in cancers. J Mol Biol. 2017;429:1978–93.
Sohier P, Luscan A, Lloyd A, Ashelford K, Laurendeau I, Briand-Suleau A, et al. Confirmation of mutation landscape of NF1-associated malignant peripheral nerve sheath tumors. Genes Chromosomes Cancer. 2017;56:421–6.
De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, et al. PRC2 loss amplifies ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–51.
Wang G, Ye H, Wang X, Liu B. Polycomb repressive complex 2 controls cardiac cell fate decision via interacting with RNA: promiscuously or well-ordered. Front Genet. 2022;13:1011228.
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–83.
Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013;24:206–14.
Ounzain S, Micheletti R, Arnan C, Plaisance I, Cecchi D, Schroen B et al. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J Mol Cell Cardiol. 2015;89 Pt A:98–112.
Wang Z, Zhang X-J, Ji Y-X, Zhang P, Deng K-Q, Gong J, et al. The long noncoding RNA chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. 2016;22:1131–9.
Kang X, Zhao Y, Van Arsdell G, Nelson SF, Touma M. Ppp1r1b-lncRNA inhibits PRC2 at myogenic regulatory genes to promote cardiac and skeletal muscle development in mouse and human. RNA. 2020;26:481–91.
Liu J, Liu S, Han L, Sheng Y, Zhang Y, Kim I-M, et al. LncRNA HBL1 is required for genome-wide PRC2 occupancy and function in cardiogenesis from human pluripotent stem cells. Development. 2021;148:dev199628.
Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet. 2017;91:507–19.
Lee K, Fu H, Aladjem MI, Myung K. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J Cell Biol. 2013;200:31–44.
Shemesh K, Sebesta M, Pacesa M, Sau S, Bronstein A, Parnas O, et al. A structure-function analysis of the yeast Elg1 protein reveals the importance of PCNA unloading in genome stability maintenance. Nucleic Acids Res. 2017;45:3189–203.
Paul Solomon Devakumar LJ, Gaubitz C, Lundblad V, Kelch BA, Kubota T. Effective mismatch repair depends on timely control of PCNA retention on DNA by the Elg1 complex. Nucleic Acids Res. 2019;47:6826–41.
Bell DW, Sikdar N, Lee K-Y, Price JC, Chatterjee R, Park H-D, et al. Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLoS Genet. 2011;7:e1002245.
Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet. 2015;47:164–71.
Venkateswarlu K, Brandom KG, Yun H. PI-3-kinase-dependent membrane recruitment of centaurin-alpha2 is essential for its effect on ARF6-mediated actin cytoskeleton reorganisation. J Cell Sci. 2007;120:792–801.
Zuccotti P, Cartelli D, Stroppi M, Pandini V, Venturin M, Aliverti A, et al. Centaurin-α2 interacts with β-tubulin and stabilizes microtubules. PLoS ONE. 2012;7:e52867.
Venturin M, Carra S, Gaudenzi G, Brunelli S, Gallo GR, Moncini S, et al. ADAP2 in heart development: a candidate gene for the occurrence of cardiovascular malformations in NF1 microdeletion syndrome. J Med Genet. 2014;51:436–43.
Bist P, Kim SS-Y, Pulloor NK, McCaffrey K, Nair SK, Liu Y, et al. ArfGAP Domain-Containing protein 2 (ADAP2) integrates upstream and downstream modules of RIG-I Signaling and facilitates type I Interferon Production. Mol Cell Biol. 2017;37:e00537–16.
Fletcher JS, Pundavela J, Ratner N. After Nf1 loss in Schwann cells, inflammation drives neurofibroma formation. Neurooncol Adv. 2020;2(Suppl 1):i23–32.
Wegscheid ML, Anastasaki C, Hartigan KA, Cobb OM, Papke JB, Traber JN, et al. Patient-derived iPSC-cerebral organoid modeling of the 17q11.2 microdeletion syndrome establishes CRLF3 as a critical regulator of neurogenesis. Cell Rep. 2021;36:109315.
Yan X, Zheng W, Geng S, Zhou M, Xu T. Cytokine receptor-like factor 3 negatively regulates antiviral immunity by promoting the degradation of TBK1 in Teleost Fish. J Virol. 2023;97:e0179222.
Douglas J, Cilliers D, Coleman K, Tatton-Brown K, Barker K, Bernhard B, et al. Mutations in RNF135, a gene within the NF1 microdeletion region, cause phenotypic abnormalities including overgrowth. Nat Genet. 2007;39:963–5.
Visser R, Koelma N, Vijfhuizen L, van der Wielen MJR, Kant SG, Breuning MH, et al. RNF135 mutations are not present in patients with Sotos syndrome-like features. Am J Med Genet A. 2009;149A:806–8.
Tastet J, Decalonne L, Marouillat S, Malvy J, Thépault R-A, Toutain A, et al. Mutation screening of the ubiquitin ligase gene RNF135 in French patients with autism. Psychiatr Genet. 2015;25:263–7.
Jin J, Zhao L, Li Z. The E3 ubiquitin ligase RNF135 regulates the tumorigenesis activity of tongue cancer SCC25 cells. Cancer Med. 2016;5:3140–6.
Martin I, Andres CR, Védrine S, Tabagh R, Michelle C, Jourdan M-L, et al. Effect of the oligodendrocyte myelin glycoprotein (OMgp) on the expansion and neuronal differentiation of rat neural stem cells. Brain Res. 2009;1284:22–30.
Oliveira AF, Yasuda R. Neurofibromin is the major ras inactivator in dendritic spines. J Neurosci. 2014;34:776–83.
Piddubnyak V, Rigou P, Michel L, Rain J-C, Geneste O, Wolkenstein P, et al. Positive regulation of apoptosis by HCA66, a new Apaf-1 interacting protein, and its putative role in the physiopathology of NF1 microdeletion syndrome patients. Cell Death Differ. 2007;14:1222–33.
Kehrer-Sawatzki H, Mautner V-F, Cooper DN. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet. 2017;136:349–76.
Salvi A, Conde I, Abeni E, Arici B, Grossi I, Specchia C, et al. Effects of miR-193a and sorafenib on hepatocellular carcinoma cells. Mol Cancer. 2013;12:162.
Yang Y, Zhou L, Lu L, Wang L, Li X, Jiang P, et al. A novel miR-193a-5p-YY1-APC regulatory axis in human endometrioid endometrial adenocarcinoma. Oncogene. 2013;32:3432–42.
Yu T, Li J, Yan M, Liu L, Lin H, Zhao F, et al. MicroRNA-193a-3p and– 5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene. 2015;34:413–23.
Tsai K-W, Leung C-M, Lo Y-H, Chen T-W, Chan W-C, Yu S-Y, et al. Arm selection preference of MicroRNA-193a varies in breast Cancer. Sci Rep. 2016;6:28176.
Venturin M, Bentivegna A, Moroni R, Larizza L, Riva P. Evidence by expression analysis of candidate genes for congenital heart defects in the NF1 microdeletion interval. Ann Hum Genet. 2005;69:508–16.
Bernardinelli Y, Nikonenko I, Muller D. Structural plasticity: mechanisms and contribution to developmental psychiatric disorders. Front Neuroanat. 2014;8:123.
Descheemaeker MJ, Roelandts K, De Raedt T, Brems H, Fryns JP, Legius E. Intelligence in individuals with a neurofibromatosis type 1 microdeletion. Am J Med Genet A. 2004;131:325–6.
Vogt J, Nguyen R, Kluwe L, Schuhmann M, Roehl AC, Mußotter T, et al. Delineation of the clinical phenotype associated with non-mosaic type-2 NF1 deletions: two case reports. J Med Case Rep. 2011;5:577.
Lemay J-F, Herbert AR, Dewey DM, Innes AM. A rational approach to the child with mental retardation for the paediatrician. Paediatr Child Health. 2003;8:345–56.
Landa RJ. Efficacy of early interventions for infants and young children with, and at risk for, autism spectrum disorders. Int Rev Psychiatry. 2018;30:25–39.
Riva P, Corrado L, Natacci F, Castorina P, Wu BL, Schneider GH, et al. NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am J Hum Genet. 2000;66:100–9.
Riccardi VM. Neurofibromatosis type 1 is a disorder of dysplasia: the importance of distinguishing features, consequences, and complications. Birth Defects Res. 2010;88:9–14.
Acknowledgements
We thank all the patients and their families, clinical geneticists, and Ian Owen Fund for their support.
Funding
The authors declare no funding.
Author information
Authors and Affiliations
Contributions
E.P., L.P., and M.U. wrote the main manuscript text and L.P. prepared Fig. 1. L.P., M.G., S.K., G.S., V.V.,M.Y., and N.T. acquired the data and carried out the experiments. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols were approved by the local hôpital Cochin ethic committee (CPP17/79, A0296746, and 2015-08-11DC). Informed consent was obtained from all subjects and/or their legal guardian(s).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pacot, L., Girish, M., Knight, S. et al. Correlation between large rearrangements and patient phenotypes in NF1 deletion syndrome: an update and review. BMC Med Genomics 17, 73 (2024). https://doi.org/10.1186/s12920-024-01843-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01843-5