
Abstract
Hearing loss is the most common sensory deficit in humans, affecting 1 in 500 newborns. Due to its genetic heterogeneity, comprehensive diagnostic testing has not previously been completed in a large multiethnic cohort. To determine the aggregate contribution inheritance makes to non-syndromic hearing loss, we performed comprehensive clinical genetic testing with targeted genomic enrichment and massively parallel sequencing on 1119 sequentially accrued patients. No patient was excluded based on phenotype, inheritance or previous testing. Testing resulted in identification of the underlying genetic cause for hearing loss in 440 patients (39 %). Pathogenic variants were found in 49 genes and included missense variants (49 %), large copy number changes (18 %), small insertions and deletions (18 %), nonsense variants (8 %), splice-site alterations (6 %), and promoter variants (<1 %). The diagnostic rate varied considerably based on phenotype and was highest for patients with a positive family history of hearing loss or when the loss was congenital and symmetric. The spectrum of implicated genes showed wide ethnic variability. These findings support the more efficient utilization of medical resources through the development of evidence-based algorithms for the diagnosis of hearing loss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hearing loss is the most common sensory deficit in humans. It is diagnosed in 1 in 500 newborns and affects half of all octogenarians (Fortnum et al. 2001; Morton and Nance 2006). Although causality is multifactorial, in developed countries, a large fraction of hearing loss is genetic and non-syndromic, i.e., not associated with other phenotypes (Marazita et al. 1993). Non-syndromic hearing loss (NSHL) mimics are syndromic forms of hearing loss that present as NSHL early in life with syndromic features developing later. Type 1 Usher syndrome, for example, is an NSHL mimic presenting as congenital profound hearing loss with delayed motor milestones. The associated progressive vision loss begins in late childhood (Smith et al. 1994).
Genetic diagnosis of NSHL and NSHL mimics is valuable. It provides prognostic information on possible progression of hearing loss, permits meaningful genetic counseling, and impacts treatment decisions (Kimberling et al. 2010). A positive diagnosis also saves healthcare dollars by directing the clinical evaluation and obviating unnecessary testing such as the routine use of imaging. The challenge, however, is in providing comprehensive genetic testing. Hearing loss is extremely heterogeneous, with over 90 genes causally implicated in NSHL (Van Camp and Smith 2015). Although historically this heterogeneity restricted genetic testing to just a few genes (Hilgert et al. 2009), the advent of targeted genomic enrichment and massively parallel sequencing (TGE + MPS) has revolutionized the clinical care of the patient with hearing loss by making comprehensive genetic testing possible (Shearer and Smith 2015).
TGE + MPS have been used in several small cohorts with positive diagnostic rates that range from 10 to 83 % [reviewed in (Shearer and Smith 2015)]. This variability reflects selection bias (i.e., including only a select ethnicity or only patients with a positive family history for hearing loss), platform bias (i.e., including only a limited number of genes), and analytic bias (i.e., neglecting to consider copy number variations in the analysis) (Hoppman et al. 2013; Ji et al. 2014; Shearer et al. 2013, 2014b). Herein, we report the analysis of the largest patient cohort to date that has undergone comprehensive clinical genetic testing for hearing loss. Of the 1119 patients presenting for testing in our clinical diagnostic laboratory, we were able to diagnose a genetic cause of deafness in 440 persons (39 %). We show that the diagnostic rate reflects ethnicity and clinical phenotype, and ranges from 1 % in patients with unilateral hearing loss to 72 % in patients of Middle Eastern ethnicity. These results provide a foundation from which to make appropriate recommendations for the use of comprehensive genetic testing in the evaluation of patients with hearing loss.
Materials and methods
Patients
Patients included in this study were sequentially referred to the Molecular Otolaryngology and Renal Research Laboratories (MORL) for clinical genetic testing from January 2012 to September 2014. All genetic screenings were done on a custom-designed TGE + MPS panel called OtoSCOPE® (Shearer et al. 2010). Relatives of patients were not included in this analysis (each nuclear and/or extended family was represented by only the proband), but no exclusions were otherwise made based upon age, age of onset, phenotype or previous testing. All available phenotype, family history, and ethnicity data were recorded. Abnormal physical exam features were classified as described in Table S1. The Institutional Review Board of the University of Iowa approved this study, and the described research was performed in accordance with the Declaration of Helsinki.
Library preparation, sequencing and bioinformatics
TGE + MPS were completed on DNA prepared from whole blood using a Sciclone NGS workstation (PerkinElmer, Waltham, MA) for sample preparation. The testing platform was either OtoSCOPE® v4 (408 individuals) or v5 (711 individuals) which targets 66 or 89 deafness-associated genes, respectively (Table S2) using custom-designed SureDesign capture technology (Agilent Technologies, Santa Clara, CA). Each platform included all known NSHL and NSHL mimic genes at the time of design (May 2011 and November 2012, respectively). Samples were analyzed in pools of 48 samples sequenced on an Illumina HiSeq (Illumina, Inc., San Diego, CA, USA) flow cell using 100-bp paired-end reads. If pre-determined quality control values were not met, the sample was rerun, as previously described (Shearer et al. 2014b).
Data were analyzed using a local installation of the open-source Galaxy software (Blankenberg et al. 2010; Goecks et al. 2010) and a combination of several other open-source tools, including read mapping with Burrows–Wheeler Alignment (BWA) (Li and Durbin 2009), duplicate removal with Picard, local re-alignment and variant calling with GATK Unified Genotyper (McKenna et al. 2010), enrichment statistics with NGSRich (Frommolt et al. 2012), and variant reporting and annotation with custom-produced software. Copy number variant analysis was performed as described (Nord et al. 2011; Shearer et al. 2014b).
Variant interpretation
On a patient-by-patient basis, all variants were discussed in the context of phenotypic data at a weekly interdisciplinary Hearing Group Meeting that included clinicians, scientists, geneticists, genetic counselors, and bioinformaticians. Each variant’s interpretation included consideration of quality/coverage depth (QD ≥ 5), minor allele frequency (MAF) from 1000 Genomes Project Database and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server [thresholds for recessive and dominant NSHL were <0.005 (excluding GJB2 variants) and <0.0005, respectively] (Shearer et al. 2014a) conservation (GERP and PhyloP) and pathogenicity prediction annotation (including PolyPhen2, SIFT, MutationTaster and LRT), and annotation within the Deafness Variation Database (deafnessvariationdatabase.org), an in-house curated, open-access database. Based upon the decision reached at Hearing Group Meeting, result letters were generated for all patients, reporting all variants with MAF <1 % to the ordering physician. In the case of positive results [variant(s) reported as ‘pathogenic’ or ‘likely pathogenic’ based on criteria defined by the American College of Medical Genetics and Genomics (ACMG) and further refined by the MORL for NSHL] (Richards et al. 2015; Shearer et al. 2014a), clinical correlation and segregation analysis were recommended. Positive results were confirmed via Sanger sequencing prior to reporting. The majority of rare variants deemed unlikely to cause hearing loss and not previously reported to be pathogenic were categorized as Variants of Unknown Significance (VUSs).
Statistical analysis
All provided clinical and phenotypic data were recorded. Diagnostic rates were compared using the Fisher exact test (comparing a specified group to all other members of the cohort) or Chi-square test (comparing more than 2 groups), with p < 0.05 considered significant. Data were compiled using Microsoft Excel and analyzed using Prism 6 (GraphPad).
Results
Patients
1119 unrelated patients were sequentially accrued during the study period. Relations were not included; otherwise, there were no exclusionary criteria. Patient demographics were binned into broad key categories: inheritance, onset, severity, laterality, physical exam and previous genetic testing (Fig. 1; Table 1). No clinical information was provided on 72 patients. For all other individuals, the available clinical information was considered during Hearing Group Meeting and discussed in the context of the genetic data. The most common characteristics included: Caucasian ethnicity (49 %); young age (93 % were <18 years of age); congenital hearing loss (56 %); severe-to-profound hearing loss (36 %); and symmetric impairment (48 %). Patients most commonly had no family history of hearing loss (54 %) and a normal physical exam (61 %).

Diagnostic rates are dependent on patient-specific clinical and phenotypic characteristics and are shown as the percentage of patients with the noted characteristic. Background shading separates categories. N for each characteristic is listed after the label. Dashed line indicates the overall diagnostic rate for this study (39.3 %). Fisher exact test used to determine statistical significance with *p < 0.05 and **p < 0.005
Genetic diagnoses
We identified a genetic cause of hearing loss in 440 patients (39 %) (Table S3). Of these patients, 101 (23 %) received a genetic diagnosis implicating an NSHL mimic, which included Usher syndrome (59 patients), Pendred syndrome (29 patients), Deafness-infertility syndrome (6 males and 1 female with NSHL), Alström syndrome (1 patient), autosomal dominant non-ocular Stickler syndrome (1 patient), branchiootorenal syndrome (BOR) (2 patients), MYH9-associated disease (1 patient), and Wolfram syndrome (1 patient) (Table S4).
Panel versioning
During the course of this study, the TGE + MPS platform was updated from v4 to v5 as part of our standard operating procedure, increasing the number of genes screened from 66 to 89. Of the 711 patients analyzed on v5, 11 patients carried causative variants in genes not included in v4, thus increasing the diagnostic rate by 2 % in all patients screened with V5 and accounting for 4 % of all positive diagnoses (11 of 263 positive diagnoses). Read metrics for V4 and V5 are shown in Table S5. Although patients sequenced with v5 had a lower average number of reads and lower average target coverage, the percentage of reads overlapping target was higher, as was the coverage at 1, 20, and 30×.
Variant identification
Our analysis of 1119 patients identified 5900 variants, which we reported to healthcare providers. 14 % of variants were considered causally related to the hearing loss phenotype and reported as pathogenic or likely pathogenic; 4 % were previously reported pathogenic variants for recessive hearing loss, with a second variant not identified (carrier status); and 82 % of variants were reported as VUSs. The median number of reported variants was 4 (range = 0–14) and 5 (0–19) for v4 and v5, respectively (Fig. S1).
Diagnostic rate and phenotype
There was considerable phenotypic diversity that impacted the overall diagnostic rate of 39 % (Fig. 1). In patients with a family history of dominant hearing loss, for example, the diagnostic rate was 50 % (p < 0.05), while in patients with a family history of recessive hearing loss it was only 41 % (not significant—n.s.). In patients with no family history of hearing loss, the diagnostic rate was 37 % (p < 0.05).
When age of onset is considered, patients with congenital hearing loss had a diagnostic rate of 44 %, which was significantly greater than the diagnostic rate in patients with childhood (29 %)- or adult (28 %)-onset hearing loss (p < 0.005 in both cases). Patients with bilateral hearing loss were significantly more likely to receive a diagnosis than patients with asymmetric or unilateral hearing loss (44, 22 and 1 %, respectively; p < 0.005). Patients with conductive or mixed hearing loss had a decreased likelihood of receiving a genetic diagnosis (17 and 21 %, respectively), but the difference was not significant.
Any kind of abnormality on physical exam decreased the likelihood of a genetic diagnosis using this panel (27 %, p < 0.005), as compared to patients with NSHL (42 %, n.s.). In patients with a clinical diagnosis of Usher or BOR syndromes, the diagnostic rate was 31 and 37 %, respectively. In none of the 15 patients with neurological findings (seizures or severe mental retardation) and hearing loss was a non-syndromic genetic cause for deafness identified (Table S6).
Combining demographic characteristics provided a more realistic assessment of the diagnostic rate (Figs. 1, 2). Patients with dominant, recessive or no family history of hearing loss had diagnostic rates of 50, 41, and 37 %, respectively. If the hearing loss was also congenital, the diagnostic rate increased to 55, 43, and 44 %. Additional phenotypic characteristics further improved the diagnostic rate (Fig. S2). For example, a patient with a negative family history for hearing loss had a lower-than-average diagnostic rate (37 %); however, if the hearing loss was congenital, the diagnostic rate increased to 44 % (p < 0.005 as compared to patients with non-congenital hearing loss and a negative family history for hearing loss). With congenital onset and symmetric hearing loss, the rate increased to 48 % (p < 0.005), and if the physical examination was normal, it increased further to 51 % (p < 0.005). The same trend was true for patients with family histories of dominant and recessive hearing loss—their diagnostic rates jumped to 67 and 55 %, respectively, when the hearing loss was congenital and symmetric and the physical examination was otherwise normal.

Diagnostic rate is influenced by ethnic, clinical and phenotypic characteristics. a N for each combination of two reported characteristics for all combinations. Color/shading reflects the number of patients with the paired criteria, up to the maximum of n = 683. b Diagnostic success for each corresponding category in a. Coloring/shading indicative of diagnosis: light orange indicates below average diagnostic rate, yellow indicates close to average diagnostic rate (39.3 %), and dark green indicates above average diagnostic rate. Empty squares had fewer than 10 individuals. AD autosomal dominant, AR autosomal recessive, PE physical exam, DFNB1 prior genetic DFNB1 (GJB2) testing, DFNB1 & other prior genetic testing including DFNB1 and other tests, other testing prior genetic testing excluding DFNB1 testing
For adult-onset hearing loss, the diagnostic rate was 28 %, however, if the family history was positive, the diagnostic rate climbed to 50 %, and if the patient also had symmetric hearing loss, the diagnostic rate jumped again to 67 %.
Only when the hearing loss was unilateral was there a marked negative impact on diagnostic rate (1 % of patients). This finding, when combined with any other characteristic, decreased diagnostic success (Fig. 2).
Diagnostic rate by ethnicity
Ethnic differences impacted the diagnostic rate (p < 0.005). In the cohort self-identified as Caucasian (549, 49 %), the diagnostic rate was 38 %. However, in cohorts self-identified as Asian (40, 4 %) and Middle Eastern (25, 2 %), the diagnostic rate was 63 and 72 %, respectively (p < 0.005). The diagnostic rate was lowest in African Americans (51, 5 %), at 26 %, p < 0.05 (Fig. 3).

Solve rate and implicated genes across ethnicities. The 10 genes with ≥10 diagnosis for the entire cohort are plotted individually; all other genes diagnosed are grouped as “other”. Ethnic-specific differences are readily apparent
Genetic spectrum
In total, 49 genes were causally implicated in hearing loss (Table 2). However, nearly three-fourths of all diagnoses (317 of 440, 72 %) were attributable to 10 genes. The four genes most frequently implicated were GJB2 (22 %), STRC (16 %), SLC26A4, (7 %) and TECTA (5 %), although this list varied based on degree of hearing loss. For example, while variants in GJB2 were the most common cause of severe-to-profound hearing loss (20 %), STRC accounted for 30 % of diagnoses in persons with mild-to-moderate hearing loss, followed closely by GJB2 (25 %) and then TECTA (7 %). SLC26A4 pathogenic variants were identified in 7 % of patients with positive diagnoses; however, all of these patients had severe-to-profound hearing loss (10 % of severe-to-profound hearing loss).
Frequency of causative genes also varied by ethnicity (Fig. 3, S4). For example, amongst self-identified Caucasian and Hispanics, STRC-related deafness was just as likely to be diagnosed as GJB2-related deafness (21 vs. 20 % and 16 vs. 14 %, respectively), but in Middle Eastern or Asian patients, GJB2 diagnoses were more common than STRC diagnoses (17 vs. 6 % and 36 vs. 4 %, respectively). No African American patients were diagnosed with GJB2-related hearing loss (Fig. 3, S4).
Causal variants
The profile of causal variant type differed with inheritance pattern. Amongst all 440 diagnoses, 49 % were due to missense variants (Table S7); however, if the hearing loss was dominantly inherited, missense variants were diagnosed 85 % of the time, as compared to 46 % with recessive inheritance. Variants predicting null alleles were much more common with recessive diagnoses—CNVs, indels, nonsense variants, and splice variants made up 20, 19, 9, and 6 % of recessive and 2, 3, 5, and 5 % of dominant diagnoses. 146 CNV alleles in 9 different genes were identified as causative in 88 patients (GJB2, MYH9, OTOA, PCDH15, SLC26A4, STRC, TMC1, TMPRSS3, USH2A). These genes contributed to 20 % of all 440 diagnoses, including one dominant diagnosis.
Discussion
Amongst studies of genetic hearing loss, this report is unique as no restrictive criteria were imposed on patient selection. Comprehensive genetic testing was completed on 1119 sequentially accrued and unrelated patients. Following a collaborative diagnostic meeting (Hearing Group) at which identified genetic variants in each patient were discussed in the context of the patient-specific phenotype, a genetic cause of hearing loss was identified in 440 patients (39 %) (Table S3). Several smaller studies have reported similar diagnostic rates (Shearer and Smith 2015).
Our data show that a focused history and physical examination can guide the expected outcome when genetic testing is ordered. The phenotypic correlations that improve or decrease the diagnostic utility of genetic testing are intuitive and logical. For example, we found that a family history positive for hearing loss improved diagnosis (44 % for dominant or recessive family history compared to 37 % for no family history).
Symmetry of hearing loss also impacted diagnosis. In patients with an otherwise normal physical exam, if the hearing loss was symmetric, the diagnostic rate was 48 %. However, a genetic cause was never identified in patients with ‘presumed’ unilateral NSHL suggesting that this condition does not exist (Figs. 1, 2). In fact, the only instance of a positive genetic diagnosis associated with unilateral hearing loss was in a patient with a family history of BOR syndrome caused by a truncating variant in EYA1, a well-recognized phenotype–genotype association (Chang et al. 2004; Chen et al. 1995).
Ethnicity impacted diagnostic rate. Nearly half (49 %) of the patients in this study self-identified as Caucasian and had a diagnostic rate of 38 %. In patients of Middle Eastern ethnicity, the diagnostic rate was higher (72 %), an increase that reflects the higher coefficient of inbreeding in this population (Najmabadi and Kahrizi 2014). Coefficient of inbreeding is known to vary across populations, ranging from 0.0365 in Bedouins to 0.0026 in Japanese and 5.96E−8 in an Afro-European admixed population of Chicago (Pemberton and Rosenberg 2014).
That the diagnostic rate was lowest in African Americans and the ‘Other’ group (which included patients of African, Bahaman or Native American heritage) suggests that there is a ‘discovery gap’ to fill in these ethnic groups (Gasmelseed et al. 2004; Shan et al. 2010). Nevertheless, in all ethnic groups, a relatively large number of less frequently implicated genes accounted for 10–15 % of diagnoses (Fig. 3), implying that across populations a similar proportion of hearing loss is due to multiple, rare, ethnic-specific variants that arise randomly and independently.
In many of the world’s populations, variants in GJB2 are the predominant cause of congenital severe-to-profound ARNSHL (Kenneson et al. 2002). In this study, they accounted for 22 % of all diagnoses and 26 % of diagnoses in the congenital severe-to-profound ARNSHL cohort. The ethnic-specific breakdown of GJB2-related hearing loss in Caucasian, Hispanic, African American, Asian, and Middle Eastern patients was 20, 14, 0, 36 and 17 %, respectively (Fig. 3, S2). When corrected for GJB2 pre-screening, the percentages increased slightly (22, 16, 0, 45, and 17 %, respectively), which is in agreement with other reports (Bazazzadegan et al. 2012; Dai et al. 2009; Du et al. 2014; Pandya et al. 2003; Usami et al. 2012).
STRC causative variants accounted for 30 % of diagnoses in patients with mild-moderate hearing loss, providing the most common diagnosis among those with this degree of hearing loss. In aggregate, 16 % of diagnoses implicated STRC. It is noteworthy that the majority of causative mutations in STRC involved large CNVs (99 %), underscoring the requirement that all comprehensive genetic testing panels for hearing loss include CNV detection.
Of variants with a MAF of <0.01, the largest majority were of unknown significance (VUSs, Fig. S1). In addition, however, we identified several known or likely pathogenic variants associated with ARNSHL in genes without a second causal variant. For example, 151 of the 679 patients, in whom a genetic diagnosis was not made, carried reported ARNSHL-causal variants without having a second variant in the coding sequence of that gene. This carrier rate of 22 % is roughly 8 times higher than that reported in hearing control populations and suggests that many of these patients have yet-to-be-identified non-coding mutations (Green et al. 1999).
Variant annotation is a dynamic process. Interpretation of variants as pathogenic, likely pathogenic, VUS, likely benign and benign is continuously refined based on increasingly robust data. The Deafness Variation Database (deafnessvariationdatabase.org) captures this area of active study in an open-source, continuously updated, interpretational database that we maintain on all variant positions interrogated on the OtoSCOPE platform.
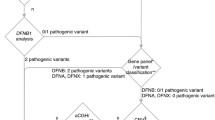
In summary, we believe that comprehensive genetic testing is a foundational diagnostic test that allows healthcare providers to make evidence-based decisions in the evaluation of hearing loss thereby providing better and more cost-effective patient care (Fig. 4, Table S8). While only 10 genes accounted for 72 % of diagnoses, 49 genes were identified as causative and 20 % of diagnoses involved at least one CNV (Table 2 and Shearer et al. (2014b)), mandating comprehensive TGE + MPS and thorough data analysis. While whole exome sequencing (WES) is becoming cheaper and for many indications more practical, a focused deafness-specific panel continues to offer the advantages of better coverage of targeted regions, greater facility to detect multiple variant types (including CNVs and complicated genomic rearrangements), substantially lower costs, higher throughput, simpler bioinformatics analysis, and focused testing, obviating the need to deal with secondary/incidental findings that otherwise inevitably arise with WES.

Recommended diagnostic workflow of a patient with hearing loss showing the value of comprehensive genetic testing (CGT) with TGE and the expected diagnostic rate in percentage. A thorough physical and history is essential and determine the expected outcome of CGT. Patients with complex phenotypes may require referral to specialists. Additional phenotypic information on select syndromes is presented in Table S6. Questions regarding the appropriateness of testing can be sent to morl@uiowa.edu. PE physical exam, CGT comprehensive genetic testing, NSHL non-syndromic hearing loss, TFT thyroid function test. aSeveral forms of syndromic hearing loss may present as NSHL and are referred to as ‘NSHL mimics’. CGT includes the diagnosis of these NSHL mimics. bCommon syndromes that can be detected by an otolaryngologist and are targeted by this CGT include Usher syndrome, Pendred syndrome and BOR syndrome. For a complete list of syndromes included on the current CGT panel see Table S8. cSome individuals will present with extremely rare/private syndromes or phenotypes that reflect the co-occurrence of two (or rarely more) syndromes. CGT should be considered for the latter cohort of patients. CGT with the OtoSCOPE panel is not indicated in patients with neurological findings such as epilepsy, intellectual delay and autism, and in patients with complex multisystem syndromes that include hearing loss caused by genes NOT targeted for capture by OtoSCOPE
References
Bazazzadegan N et al (2012) The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss–a twelve year study. Int J Pediatr Otorhinolaryngol 76:1164–1174. doi:10.1016/j.ijporl.2012.04.026
Blankenberg D et al. (2010) Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol 10:11–21. doi:10.1002/0471142727.mb1910s89 (Chapter 19:Unit 19)
Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoort VS, Schwartz CE, Smith RJ (2004) Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat 23:582–589. doi:10.1002/humu.20048
Chen A et al (1995) Phenotypic manifestations of branchio-oto-renal syndrome. Am J Med Genet 58:365–370. doi:10.1002/ajmg.1320580413
Dai P et al (2009) GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med 7:26. doi:10.1186/1479-5876-7-26
Du W, Wang Q, Zhu Y, Wang Y, Guo Y (2014) Associations between GJB2, mitochondrial 12S rRNA, SLC26A4 mutations, and hearing loss among three ethnicities. Biomed Res Int 2014:746838. doi:10.1155/2014/746838
Fortnum HM, Summerfield AQ, Marshall DH, Davis AC, Bamford JM (2001) Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ 323:536–540
Frommolt P et al (2012) Assessing the enrichment performance in targeted resequencing experiments. Hum Mutat 33:635–641. doi:10.1002/humu.22036
Gasmelseed NM et al (2004) Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum Mutat 23:206–207. doi:10.1002/humu.9216
Goecks J, Nekrutenko A, Taylor J, Galaxy T (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86. doi:10.1186/gb-2010-11-8-r86
Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ (1999) Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA 281:2211–2216
Hilgert N, Smith RJ, Van Camp G (2009) Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res 681:189–196. doi:10.1016/j.mrrev.2008.08.002
Hoppman N, Aypar U, Brodersen P, Brown N, Wilson J, Babovic-Vuksanovic D (2013) Genetic testing for hearing loss in the United States should include deletion/duplication analysis for the deafness/infertility locus at 15q15.3. Mol Cytogenet 6:19. doi:10.1186/1755-8166-6-19
Ji H, Lu J, Wang J, Li H, Lin X (2014) Combined examination of sequence and copy number variations in human deafness genes improves diagnosis for cases of genetic deafness. BMC Ear Nose Throat Disord 14:9. doi:10.1186/1472-6815-14-9
Kenneson A, Van Naarden Braun K, Boyle C (2002) GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med 4:258–274. doi:10.1097/00125817-200207000-00004
Kimberling WJ et al (2010) Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet Med 12:512–516. doi:10.1097/GIM.0b013e3181e5afb8
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. doi:10.1093/bioinformatics/btp324
Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE (1993) Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet 46:486–491. doi:10.1002/ajmg.1320460504
McKenna A et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi:10.1101/gr.107524.110
Morton CC, Nance WE (2006) Newborn hearing screening–a silent revolution. N Engl J Med 354:2151–2164. doi:10.1056/NEJMra050700
Najmabadi H, Kahrizi K (2014) Genetics of non-syndromic hearing loss in the Middle East. Int J Pediatr Otorhinolaryngol 78:2026–2036. doi:10.1016/j.ijporl.2014.08.036
Nord AS, Lee M, King MC, Walsh T (2011) Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom 12:184. doi:10.1186/1471-2164-12-184
Pandya A et al (2003) Frequency and distribution of GJB2 (connexin 26) and GJB6 (connexin 30) mutations in a large North American repository of deaf probands. Genet Med 5:295–303. doi:10.1097/01.GIM.0000078026.01140.68
Pemberton TJ, Rosenberg NA (2014) Population-genetic influences on genomic estimates of the inbreeding coefficient: a global perspective. Hum Hered 77:37–48. doi:10.1159/000362878
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. doi:10.1038/gim.2015.30
Shan J et al (2010) GJB2 mutation spectrum in 209 hearing impaired individuals of predominantly Caribbean Hispanic and African descent. Int J Pediatr Otorhinolaryngol 74:611–618. doi:10.1016/j.ijporl.2010.03.004
Shearer AE et al (2010) Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci 107:21104–21109. doi:10.1073/pnas.1012989107
Shearer AE et al (2013) Advancing genetic testing for deafness with genomic technology. J Med Genet 50:627–634. doi:10.1136/jmedgenet-2013-101749
Shearer AE et al (2014a) Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am J Hum Genet 95:445–453. doi:10.1016/j.ajhg.2014.09.001
Shearer AE et al (2014b) Copy number variants are a common cause of non-syndromic hearing loss. Genome Med 6:37. doi:10.1186/gm554
Shearer AE, Smith RJ (2015) Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Head Neck Surg 153:175–182. doi:10.1177/0194599815591156
Smith RJ et al (1994) Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet 50:32–38. doi:10.1002/ajmg.1320500107
Usami S, Nishio SY, Nagano M, Abe S, Yamaguchi T, Deafness Gene Study C (2012) Simultaneous screening of multiple mutations by invader assay improves molecular diagnosis of hereditary hearing loss: a multicenter study. PLoS One 7:e31276. doi:10.1371/journal.pone.0031276
Van Camp G, Smith, RJH (2015) Hereditary hearing loss homepage. http://hereditaryhearingloss.org. Accessed 15 June 2015
Acknowledgments
We are grateful to the patients and families included in this study. We thank all physicians and genetic counselors for allowing us to help in the care of their patients. This work was supported by T32 GM007337 to the University of Iowa MSTP and by NIDCD RO1s DC003544, DC002842 and DC012049 to RJHS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
CMSH, AOB, AES, DLK, CJN, KLF, SSE, SBS, KTB, CAC, PTR, AEW, EABZ, DW, and HA disclose no conflict of interest. RJHS directs the MORL, which offers TGE + MPS as a clinical diagnostic test for hearing loss.
Additional information
A. O. Bierer, A. E. Shearer, and D. L. Kolbe all contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sloan-Heggen, C.M., Bierer, A.O., Shearer, A.E. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 135, 441–450 (2016). https://doi.org/10.1007/s00439-016-1648-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1648-8