
Abstract
The current pandemic (COVID-19) has made evident the need to approach pathogenicity from a deeper and more systematic perspective that might lead to methodologies to quickly predict new strains of microbes that could be pathogenic to humans. Here we propose as a solution a general and principled definition of pathogenicity that can be practically implemented in operational ways in a framework for characterizing and assessing the (degree of) potential pathogenicity of a microbe to a given host (e.g., a human individual) just based on DNA biomarkers, and to the point of predicting its impact on a host a priori to a meaningful degree of accuracy. The definition is based on basic biochemistry, the Gibbs free Energy of duplex formation between oligonucleotides and some deep structural properties of DNA revealed by an approximation with certain properties. We propose two operational tests based on the nearest neighbor (NN) model of the Gibbs Energy and an approximating metric (the h-distance.) Quality assessments demonstrate that these tests predict pathogenicity with an accuracy of over 80%, and sensitivity and specificity over 90%. Other tests obtained by training machine learning models on deep features extracted from DNA sequences yield scores of 90% for accuracy, 100% for sensitivity and 80% for specificity. These results hint towards the possibility of an operational, objective, and general conceptual framework for prior identification of pathogens and their impact without the cost of death or sickness in a host (e.g., humans.) Consequently, a reasonable prediction of possible pathogens might pave the way to eventually transform the way we handle and prepare for future pandemic events and mitigate the adverse impact on human health, while reducing the number of clinical trials to obtain similar results.
Similar content being viewed by others
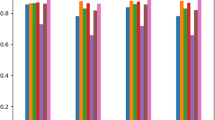
Avoid common mistakes on your manuscript.
Introduction
The COVID-19 pandemic has given a renewed sense of urgency about something we have known all along, our vulnerability to myriad strains of microbes that may cause severe damage to human health, both in individuals and communities. The overarching goal of this work is to propose a new approach to the concept of pathogenicity and to describe a framework for characterizing and assessing the degree of potential pathogenicity of a microbe to a given host (e.g., a human individual.) It is based on DNA biomarkers from both and affords a priori predictions of the impact on a host to a meaningful degree of sensitivity and specificity. To put our proposal in perspective, we first summarize the efforts made in the field of pathogenicity and immunology to address this problem.
Evolution of the concept/definition of `pathogen’
The term pathogen (borrowed from the Greek, pathos meaning disease and genos meaning origin) has been in use since the 1880s to refer to infectious microorganisms, including viruses, bacteria, protozoans, prions, viroids and fungi (Alberts et al. 2002; Casadevall and Pirofski 2014). The study of diseases caused by pathogens does not have a distinct origin but could be traced back to their documentation in Egyptian medicine, for instance, the Edwin Smith Papyrus (seventh century BC) and the Papyrus Ebers (about 1550 BC.) These records contain data regarding several bone injuries, trachoma, ulcerating lumps, parasites and other diseases (Van den Tweel and Taylor 2010). Identifying, naming and recording specific microbes that cause bodily damage (Ghosh 2017) disease or death was the first systematic attempt to narrow down the concept of pathogenicity.
Since then, several characterizations have been proposed to come to grips with the nature of pathogens. Earlier views were primarily based on microorganisms and their intrinsic properties only, although it was also implicitly understood that pathogenicity was neither invariant nor absolute (Casadevall and Pirofski 1999). In the early twentieth century, Bail proposed aggressins and Rosenow proposed virulins as microbial products ushering pathogens themselves into the host. Later in the 1900s, (Zinsser 1914; Watson and Brandly 1949) proposed grouping microorganisms into three different categories, namely, saprophytes (unable to establish themselves in living tissue), pure parasites and half parasites. Later, Falkow proposed “Molecular Koch’s Postulates” as a conceptual framework to identify the genes causing diseases (Falkow 1988) and noted that a pathogen has an intrinsic ability to breach the cell barriers of a host (Falkow 1997). These several definitions, reviewed in more detail in (Casadevall and Pirofski 1999), can be summarized as follows:
-
A microbe capable of causing disease (Hoeprich 1989; Shulman 1997).
-
A micro-organism that can grow in living tissue and produce disease (Ford 1927).
-
Any micro-organism whose survival is dependent upon its capacity to replicate and persist on or within another species by actively breaching or destroying a cellular or humoral host barrier that ordinarily restricts or inhibits other micro-organisms (Falkow 1997).
-
A parasite capable of causing or producing some disturbance in the host (Smith 1934).
These conflicting drives between host and pathogen led to an evolutionary “attack-defense” approach (Kuduva et al. 2020). Recent studies further rely on a similar approach to define pathogens. (Balloux and van Dorp 2017) defined pathogens as organisms causing diseases to their hosts, with the severity of the disease symptoms referred to as virulence. There are two types of pathogens i.e., facultative pathogens (environmental bacteria and fungi occasionally causing diseases) and obligate pathogens (requiring hosts to complete their lifecycle.) However, this definition of a pathogen remains incomplete since there are probably millions of pathogenic micro-organisms that remain unidentified. Even bioinformatic tools to detect pathogens fail to detect novel species when similar genomes are not available and have limitations related to the dependence on genome assembly or being slow to large-scale read mapping (Deneke et al. 2017).
Consequently, recent studies are solely focused on the identification of these pathogens and how they can cause diseases rather than giving a general unified and more principled definition. (Saliba et al. 2017) pointed out that understanding how bacteria cause disease requires knowledge of which genes are expressed and how they are regulated during infection. (Cosentino et al. 2013) developed a web server to predict bacterial pathogenicity based on the analysis of the input proteome, genome or raw NGS reads provided by a user. The conceptual framework behind the server had been validated using 449 sequenced bacteria with 88.6% of accuracy. (Segawa et al. 2014) argued that Matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) is an appropriate tool to diagnose rapidly and accurately to identify species that are pathogens. A case of bacterial meningitis caused by Klebsiella pneumoniae using the same method for the same purpose was also described. Similarly, (Gu et al. 2021) proposed a metagenomic next-generation sequencing test using cell-free DNA from body fluids to identify pathogens. They assessed their sensitivity/specificity at about 79%/91% for bacteria, 91% /89% for fungi, using Illumina sequencing; 75%/81% for bacteria and 91%/100% for fungi using nanopore sequencing, respectively.
Furthermore, (Liu et al. 2021) used databases of nonpathogenic and pathogenic species to make training workflows and then predict sets of pathogenic and nonpathogenic species. Random forest models used for the predictions yielded an accuracy between 88 and 93%. A comparison between metagenomic next-generation sequencing (NGS) and the conventional diagnostic methods for the detection of CNS infection in patients after allogeneic hematopoietic stem cell transplantation (allo-HSCT) was made. Thirty-eight (38) pathogens were found in 34 of 53 patients in the study (including 33 viruses, 3 bacteria and 2 fungi.) 32 pathogens were detected by mNGS and conventional testing both and 6 by mNGS only (then, those 6 cases were verified, 5 of them had an infection.) The sensitivity of mNGS and conventional testing for diagnosing CNS infections post-transplant were 97.1% and 82.9%, respectively (P = 0.106), while the specificity of mNGS and conventional testing were 94.4% and 100%, respectively (P = 1.000).
These characterizations cannot really be regarded as logically satisfactory general definitions because they still rely on “someone dying/getting sick” to establish that some microbe is pathogenic. COVID-19 and influenza have now made evident that the ability of some microorganisms, even viruses, to cause disease depends on a specific host. Thus, recent studies are shifting their focus on the two-way relationship between host and micro-organisms in the form of pathogenicity and are even advocating to ditch the term “pathogen” (Casadevall and Pirofski 2014).
An objective and operational perspective towards a pathogenic relationship between a microbe and a host is a must to be prepared for a possible insurgence of a pandemic (like COVID-19) in the future. (Credle et al. 2021) also affirms the need of such technologies or methodologies that are quickly reconfigurable to prevent or be prepared for future crises caused by emerging threatening pathogens to human health. For the same reason, (Credle et al. 2021) proposed a method based on NGS and claimed to be able to detect new strains based on rapid analysis. A short time frame is very useful for surveillance and can separate targeted from untargeted RNA molecules (especially when the spread of a disease is in its early phase.)
All these considerations point towards a single clear conclusion, that although there are several approaches to make a distinction between pathogens and nonpathogens, there is no general, principled, and operational definition of pathogens. The purpose of this paper is to propose such a general definition of a pathogenic relationship between two biological organisms. Next, we present the first proposal for such a definition. In the "Materials and methods" section, we present the description of the conceptual framework necessary to implement it in a practical way. We then describe the datasets used to validate and assess its quality. In the "Discussion and conclusion" section, we present the results of the assessment and summarize our findings, along with some advantages, limitations and conclusions.
A principled computational definition of ‘pathogenicity’
To find a solution to the problem of identifying the pathogenicity of a microbe, one should follow a wholistic rather than a reductionist approach. There are several factors involved for a microbe to cause disturbances in the homeostasis of a host. First, a microbe is a changing entity once it enters a host. Second, a host likewise changes because of infection. Third, they interact with each other (i.e., host response to a pathogen’s attack has also some impact on the latter.) Fourth, both parties interact with each other in an uncontrolled dynamic environment (not in a wet lab where factors like temperature, pressure and so on are closely monitored and controlled.) This simply implies that the term ‘pathogen’ is, like motion, relative and does not make absolute sense. Therefore, the right approach is to focus rather on the nature of the relationship between a microbe and a host.
Definition 1.
A specimen P has a pathogenic relationship with a species H over a given period of time, if and only if.
-
P interacts with any specimen in H and begins to reproduce.
-
H produces a defense in response to counteract the resulting colony of Ps.
-
Ps may push back, and H may counteract, until H reaches a stable condition that may be different from the condition prior to interaction with P.
-
These three conditions remain true with at least K other specimens in H, in the absence of any other such P*, for an appropriate value of K (e.g., 32.)
There are situations where two pathogens can attack the host simultaneously and may be successful jointly, but not individually. Therefore, a general definition should allow for multi-way pathogenic relationships. However, in this first study, we will only address in-depth binary relationships.
Materials and methods
Therefore, in this approach, the problem of identifying whether a given micro-organism is pathogenic to a human or not can be approached at first as a binary classification problem into the categories of pathogen/nonpathogen. To solve it using DNA samples alone, we identified a proxy DNA sequence to represent a microbe and another proxy sequence to represent the individual Homo sapiens host. The primary limitation in gathering the data was their availability, including labels (pathogenic/non) for the genomic sequences (paired data.) The methods and tests described below can in principle be applied to any organism in the given taxa (bacteria and fungi.) The sequences required to obtain such proxies for these labeled microbes were downloaded from GenBank (Benson et al. 2012), as shown in tables in the Appendix. Custom-made MATLAB and Python scripts were used to run the tests stated below and compute some standard metrics for the assessment of the quality of the proposed definition.
The key idea behind our proposal is to obtain a pattern of hybridization between the proxy sequences from micro-organisms and host on a certain set of oligos (the so-called grid below.) Once such a pattern is obtained, then a comparison is made (either by an analytic or a machine learning model) to determine the pathogenicity of the micro-organisms in reference to the host. The precise proposed definition of pathogenicity is given next.
The PNP-G test based on Gibbs free energies
We selected a so-called grid from the host as a proxy to obtain a pattern of hybridization affinity between a host and a micro-organism, as determined by a model of hybridization (Gibbs Energy or h-distance) (Azizzadeh et al. 2021). The grid consists of a number m of n-mers selected randomly from a consensus sequence (representing 32 specimens of Homo sapiens) with a uniform distribution. The critical parameter that turned out to be most helpful in the classification is the count of the number of hybridizations of the proxies of such microbes with the grids consisting of m = 100 or 200 genomic fragments of length n = 20 or 40. The test is determined by two parameters ε and r, selected as optimal thresholds with radius defining the upper and lower bounds for the count of hybridizations at 37 \(^\circ\) Celsius to gene fragments in the grid. (Similar results can be obtained at a fever temperature of 41 \(^\circ\) C.) To optimize the choice of such parameters, we quantized the range of possible values (between 1 and 65 000 with increment steps of 1 for both Gibbs energy and h-distance) and computed the corresponding accuracy, specificity, and sensitivity of the test for values of upper and lower bounds such that the average of these bounds ε and r is the positive difference between these bounds and the average on the data (described below.) The hybridization energy threshold for hybridization with Gibbs energy was assumed to be τ = − 6 kcal/mol as it is usually assumed, i.e., two oligonucleotides (of length at most 60) hybridize if the Gibbs energy (as given by the Nearest-Neighbor model (SantaLucia 1998)) of the pair is at most − 6 kcal/mol. The test can thus be stated as follows.
The pathogenicity Gibbs test PNP-G (n, m, ε, r)
Pre-Conditions: a `grid’ Γ consisting of m randomly selected n-mers y with a uniform distribution from a host H. Thresholds τ and ε for hybridization to n-mers in Γ.
Input: two sets of n-mers M (a microbe) and H (a host).
Output: `Pathogenic’ or `Nonpathogenic’.
Procedure: If the count of the number of hybridizations between x in M and z in Γ is in the interval [ε – r, ε + r], return `pathogenic’; else return ‘nonpathogenic’.
To estimate the Gibbs energy in the metric equivalent, we used the h-distance metric approximation of the Gibbs energy introduced in (Garzon et al. 1997) to perform a similar test PNP-h test with hybridization threshold τ = 14 (bacteria) and τ = 27 (fungi) and ε = 19,460.5 (bacteria) and ε = 59,271.5 (fungi) with r = 123.5 (bacteria) and r = 101.5 (fungi), for all performance scores>=80% \(\ge 80\%\). The PNP-G test appears to be more accurate. The parameters used in both tests are shown in Tables 1 and 2.
Machine learning models using features obtained from Nxh bases
In biology, microarrays used to be the first choice for mining huge amounts of data from DNA sequences (Schena 2003). But noise introduced by hybridization uncertainty in the readout analyses and the subjective selection of relevant features (to be used as targets on the chip) have made this type of analysis irreproducible and hence unreliable. The recent advances in next-generation sequencing and bioinformatic tools to analyze these sequences have addressed these issues to some extent (Roh et al. 2010). Our new approach also relies on hybridization affinity, but some deep structure revealed by analyses of the hybridization landscapes in the h-distance model affords designs of so-called nxh chips (or bases) based on a more principled and objective foundation. We summarize them here briefly to make this paper self-contained. (The reader is referred to (Garzon and Bobba 2012; Garzon and Mainali 2017; Garzon and Mainali 2021) for more details.)
A noncrosshybridizing (nxh) basis can be defined as a set consisting of pairs of Watson–Crick (WC) complementary oligonucleotides of fixed length (so-called pmers) that do not hybridize to one another under certain reaction conditions specified by a parameter τ > 0. The following property summarizes the ideal requirements for an ideal set of such oligonucleotides (Garzon and Minali 2017).
(1) A nxh basis should consist of a sufficient number of paired oligonucleotides (hereafter pmers) in such a way that every random n-mer of the same length n in any target sequence(s) will hybridize with exactly one of them (as determined by a stringency threshold \(\tau\).)
With the proper design of an nxh basis in hand, a custom python script was written to reduce arbitrarily long DNA sequences x to a few but very informative features in a numerical vector. First, we split a proxy DNA sequence x into fragments of the same length matching that of oligos in the basis. Next, based on the hybridization affinity between each fragment and the oligos on the basis (hereafter referred to as probes), we counted the number of fragments in the sequence that are likely to hybridize with each probe, according to the given hybridization criterion. Once the counts for the probes in the basis are obtained, we normalized the vector containing these counts using a partition function so that the sum of all normalized counts adds up to 1. We use the term genomic signature to refer to such a normalized vector. The decision about a possible hybridization is made based on a metric approximation of Gibbs energy between any two oligos, known as hybridization (h-) distance (Garzon et al. 1997) quantifying the likelihood of hybridization between such oligos. In other words, for any two oligos of the same length, if their h-distance is 0, then they are either identical or WC complements and they are most likely to hybridize. On the other hand, if their h-distance is maximum (i.e., their length n), then they are least likely to hybridize (e.g. pmers aaa/ttt and ccc/ggg). Furthermore, (Garzon and Bobba 2012) showed that hybridization decisions under a set of reaction conditions made based on a criterion that h-distance between any two oligos be less than \(\tau\)(for a suitable choice of threshold reflecting reaction conditions, such as temperature and ph), agree at least 80% of the time with decisions made using the nearest neighbor model of Gibbs energy for hybridization affinity, assuming a threshold of Gibbs energy less than \(\tau\) = − 6 kcal/mol for hybridization.
Once the sequences were transformed into genomic signatures, they were fed to machine learning models (as described in Table 3) to differentiate between pathogenic and nonpathogenic microorganisms.
Data and assessment
To assess the quality of the tests and the definition of pathogenicity, we selected three sets of DNA sequences and downloaded them from GenBank (Benson et al. 2012). The first contained coding sequences of whole genomes in 25 known pathogenic and 25 known nonpathogenic bacteria for Homo sapiens in general, according to (Cosentino et al. 2013). A similar selection was made for a second sample of fungi (CDC 2014). The third sample consisted of four mitochondrial genes (COI, COII, COIII, CytB) in 32 specimens of Homo sapiens (as a host species; the specimens contained in these samples are fully referenced in the Appendix.) As mentioned above, no further specific criteria were used in choosing the organisms other than the availability of their labels about their pathogenicity (pathogenic/non) so that paired data could be used to assess the quality of the tests below. In particular, whether these bacteria are present in human hosts is unknown, although it is fairly safe to assume that microbes pathogenic to a given healthy host are likely to be alien to that host and that no horizontal gene transfers have occurred from the microbe.
The proxies of these organisms to be used in the analyses were chosen as follows. For a proxy for a microbe bacterium specimen, we randomly (uniform distribution) selected m = 200 or 300 random n-mers with n = 20 as well as 40 from the genomic sequence of the specimen. The same procedure was repeated to obtain four datasets for fungi. Thus, we obtained eight (8) datasets altogether. Similarly, to obtain a proxy for a grid G for the species host Homo sapiens, we created a pool of all n-mers (when n = 20 or 40) from all specimens in our sample and then selected m = 100 as well as 200 n-mers randomly under the uniform distribution. Finally, the Gibbs Energies (SantaLucia 1998) and h-distances (Garzon et al. 1997; Garzon and Bobba 2012; Garzon and Mainali 2021) between every pair of oligomers from each pair of microbe and host proxies were computed. The detailed description of the proxies is shown in Table 4.
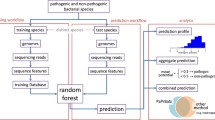
We also trained ML models (as shown in Table 3) using genomic signatures of sequences in each sample using the workflow described above in Fig. 1 on the nxh bases in Table 5. These models were trained on three samples containing bacteria only, fungi only and both together, using 80% of each data set for training. Then these models were used to make predictions about the pathogenicity of these microorganisms on the remaining testing dataset (the remaining points for testing 20% of the full dataset.)

A DNA sequence x is shredded into fragments of the same length n as that of the probes on an nxh basis so that the total number of fragments hybridizing with each oligo can be counted for each probe to obtain a feature vector from x. The oligos for the basis are judiciously selected in such a way that no cross hybridization occurs among probes in the basis itself and, moreover, that every random fragment hybridizes to (ideally exactly) one probe. An ideal basis thus produces feature vectors that are fully reproducible and contain much of the information in the original sequence x
The metrics for assessment are standard in the medical field (for example, for diagnostics tests.) Quantitative metrics are used because it is important to quantify the accuracy of the tests and make an approximation of the extent to which they discriminate between the two conditions (Šimundić 2009) and because they might afford a finer distinction between degrees of pathogenicity. Accuracy describes “the proportion of all tests that was correctly predicted” (Lutgendorf and Stoll 2016). Two other measures give finer information about the accuracy of the tests and quantify their discriminative potential, sensitivity and specificity. These are complementary, with the first one quantifying the ability of the test to identify the portion of true positives of the total of evaluations (Šimundić 2009); the higher the sensitivity, the fewer false negatives are obtained (Maxim et al. 2014). On the other hand, specificity quantifies true negatives (Šimundić 2009); the higher the specificity, the fewer false positives are obtained (Maxim et al. 2014). According to the recent literature (Cosentino et al. 2013; Saliba et al. 2017; Gu et al. 2021; Liu et al. 2021), we can conclude that a model with a sensitivity above 75% and specificity above 80% could be deemed acceptable.
Results
The PNP-G test based on Gibbs energies and PNP-h test on h-distance
We present the parameters used in both tests to define pathogenic relationships between microbes and hosts in Tables 1 and 2. The results on prediction based on PNP-G and PNP-h tests are shown in Figs. 2 and 3. We report the grids with best performance scores (others were fairly scattered below them.) On the one hand, for the PNP-G test, grid hsGrid200 gives the optimal scores for the bacterial set bacs25-20C with 82% of accuracy, 100% of sensitivity and 64% of specificity. Similarly, they achieved performance scores of 98% for accuracy, 96% for sensitivity and 100% for specificity, on the grid hsGrid200 for the fungal dataset funs20C. On the other hand, we got optimal scores of 86% accuracy, 92% sensitivity and 80% specificity for bacteria and 92% of all performance scores (accuracy, sensitivity, and specificity) for fungi with the PNP-h test.

Performance assessment of the definition of pathogenicity of bacteria and fungi using thresholding methods, based on the decision about hybridization events between oligos in the proxies of a host and a microorganism (Top: based on Gibbs Energy and Bottom: based on h-distance.) The x-axis represents different data sets for proxies and grids (IDs are in Table 4.)

Performance assessment of the definition of pathogenicity of bacteria (top), fungi (middle) and combined (bottom) obtained using machine learning models trained on genomic signatures
Performance of machine learning (ML) on nxh bases
The results of prediction based on machine learning models (as shown in Table 3) are shown in Fig. 3. Again, we report models with the best performance scores since it is preferable to use only the most successful test for each case. The basis 3mE4b-2 gives the best result with the models kNN, RBF and MLP with perfect scores for bacteria. However, when we analyze these signatures using ML models to obtain the definition of pathogenicity for fungi, the scores dropped significantly. Figure 3 (middle) shows that the models DT and AB give the best performance when trained using combined features from the bases 4mP3-3 and 8mP10-4. They show a perfect score for sensitivity; moreover, the remaining scores are all above 80%. These findings are consistent with the current literature showing challenges in the definition of pathogenicity among fungi (CDC 2014). Surprisingly, the worst performing ML models were Support Vector Machines (SVM) on Radial Basis Transfer functions (RBFs.) The models were trained using combined features from all bases. Finally, for the third sample containing the combined taxa, only one model (AB) showed the best performance in predicting pathogenicity among bacteria and fungi, although the remaining models gave good scores (above 75%) for sensitivity.
Discussion and conclusion
We have proposed a novel approach to pathogenicity based on a computational characterization of the pathogenic relationship between a microbe and a host. The operational implementation of the definition yielded scores of over 80% for accuracy and over 90% for sensitivity and specificity by two PNP tests for both bacterial and fungal taxa. Tests on machine learning models on the combined taxa achieved scores about 90% for accuracy, 100% for sensitivity and 80% for specificity.
A question may arise as to whether these results are general enough to scale them to pathogens and Homo sapiens in general. To test the diversity of this dataset to ensure widespread coverage of the whole population of pathogens and hosts, we calculated the average Gibbs energies and h-distances between all pairs of specimens in the datasets (as an average of the Gibbs energy or h-distance between pairs of their 200 or 300 randomly selected 20- and 40-mers from their proxy sequences.) Their averages in Table 6 confirm that this is indeed the case. They show that hybridizations within groups are unlikely to occur on the average within each group (bacteria and fungi) and even across groups, according to the hybridization metrics used in the tests, i.e., these microbes in the datasets are biologically very different from one another.
These results have some interesting implications. First, both PNP tests include proxies of relatively small size. Particularly, coding sequences on whole genome of bacteria include millions of nucleotides in general, but our proxies include at most 12 000 nucleotides. (Ghosh 2017; Garzon and Mainali 2021; Watson and Brandly 1949). Therefore, it is interesting that the tests were able to achieve performance over 80% for bacteria and 95% for fungi in all scores on a proxy of such a small proportion of the entire genome. Second, the PNP-h test gives better accuracy and sensitivity for bacteria than the PNP-G test, even though the h-distance metric is just an approximation of Gibb’s Energy Nearest-Neighbor model (SantaLucia 1998) that allows about 20% error for hybridization decisions (Garzon et al. 1997; Garzon 2014; Garzon and Bobba 2012). Even when the former gives a lower score than the latter, the difference in the corresponding score is insignificant compared to this margin of error while making hybridization decisions when compared to the Gibbs Energy Nearest-Neighbor Model (SantaLucia 1998). Third, not all machine learning models were suitable to obtain classifiers for pathogenicity over all datasets combined. An alternative approach would be to select different models for different taxa (here bacteria and fungi.) Fourth, our proposed solutions can easily provide a more reasonable definition of pathogenicity in the case of bacteria than fungi. This fact is consistent with biological knowledge that fungi are eukaryotes and have a more complex cellular structure and physiology. It is worth noting the fact that we humans share a more diverse and versatile symbiotic relationship with bacteria. For example, fungal diversity in the human population was found to be low in largely stable colonization over time, with the occasional transient species. Moreover, a large proportion of fungi may be of dietary origin and thus may not be functionally relevant in the human gut environment (Huseyin et al. 2017). Fifth, the results from this method imply that pathogens feature a higher Gibbs energy with the genome of a host. Further, as remarked above in the ''Data Assessment" section, this affinity is not likely to be due to horizontal gene transfers between bacteria and hosts. Upon reflection, these results are consistent with the local nature of the interactions in the biological machinery of living organisms.
This line of research opens some new possibilities as well. Although in this first approach the problem of pathogenicity was addressed as a binary classification problem, our method can be readily extended to a prediction problem of the degree of pathogenicity of a microbe to a host, a reasonable next step. Furthermore, it may be possible to evaluate the degree of pathogenicity in different strains of the same species. Second, in our time, there is a growing interest in personalized medicine. These methods can be readily extended to grids for individual specimens of H. sapiens. That is an intriguing possibility for further research. Third, we only presented the definition of pathogenic relationship in relation to the human species. It would be interesting to explore how these tests perform with other hosts. Finally, we performed these experiments in silico only and have not made any analysis to carry out these tests with appropriate parameters and their results in a wet lab. It would be interesting to explore the translation of these parameters into physical experimental conditions.
Data availability
The data used in this research are publicly available at http://bmc.memphis.edu/pathos/.
References
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002) Introduction to pathogens. In Molecular biology of the cell, 4th edn. Garland Science.
Azizzadeh S, Garzon M, Mainali S (2021) Classifying single nucleotide polymorphisms in humans. Mol Genet Genomics 296:1161–1173
Balloux F, van Dorp L (2017) Q&A: What are pathogens, and what have they done to and for us? BMC Biol 15(1):1–6
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2012) GenBank. Nucleic Acids Res 41(D1):D36–D42
Casadevall A, Pirofski LA (1999) Host-pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infect Immun 67(8):3703–3713
Casadevall A, Pirofski LA (2014) Microbiology: ditch the term pathogen. Nat News 516(7530):165
Centers for Disease Control and Prevention (CDC) (2014) Water-related diseases and contaminants in public water systems.
Cosentino S, Voldby Larsen M, Møller Aarestrup F, Lund O (2013) PathogenFinder-distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 8(10):e77302
Credle JJ, Robinson ML, Gunn J, Monaco D, Sie B, Tchir A et al (2021) Highly multiplexed oligonucleotide probe-ligation testing enables efficient extraction-free SARS-CoV-2 detection and viral genotyping. Mod Pathol 34(6):1093–1103
Deneke C, Rentzsch R, Renard BY (2017) PaPrBaG: A machine learning approach for the detection of novel pathogens from NGS data. Sci Rep 7(1):1–13
Falkow S (1997) What is a pathogen? ASM News 63:359
Falkow S (1988) Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis 10(2):S274–S276
Ford WW (1927) Text-book of bacteriology. W. B. Saunders Company
Garzon MH (2014) DNA codeword design: theory and applications. Parallel Process Lett 24(02):1440001
Garzon MH, Mainali S (2021) Deep structure of DNA for genomic analysis. Hum Mol Genet 31(4):576–586. https://doi.org/10.1093/hmg/ddab272
Garzon MH, Mainali S (2017) Towards reliable microarray analysis and design. In: 9th international conference on bioinformatics and computational biology, ISCA, 6 pp.
Garzon M, Neathery P, Deaton R, Murphy RC, Franceschetti DR, Stevens Jr SE (1997) A new metric for DNA computing. In: Proceedings of the 2nd genetic programming conference, vol 32, No. 1. Morgan Kaufman, pp 636–638
Garzon MH, Bobba KC (2012) A geometric approach to Gibbs energy landscapes and optimal DNA codeword design. In: International workshop on DNA-based computers. Springer, Berlin, Heidelberg, pp 73–85
Ghosh SK (2017) Giovanni Battista Morgagni (1682–1771): father of pathologic anatomy and pioneer of modern medicine. Anat Sci Int 92(3):305–312
Gu W, Deng X, Lee M, Sucu YD, Arevalo S, Stryke D, Federman S, Gopez A, Reyes K, Zorn K, Sample H (2021) Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med 27(1):115–124
Hoeprich PD (1989) Host-parasite relationships and the pathogenesis of infectious disease. In: Hoeprich PD, Jordan MC (eds) Infectious diseases. Lippincott, Philadelphia, pp 41–53
Huseyin CE, O’Toole PW, Cotter PD, Scanlan PD (2017) Forgotten fungi—the gut mycobiome in human health and disease. FEMS Microbiol Rev 41(4):479–511
Kudva IT, Cornick NA, Plummer PJ, Zhang Q, Nicholson TL, Bannantine JP, Bellaire BH (eds) (2020) Virulence mechanisms of bacterial pathogens. Wiley
Liu W, Fan Z, Zhang Y, Huang F, Xu N, Xuan L, et al (2021) Metagenomic next-generation sequencing for identifying pathogens in central nervous system complications after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 1–6.
Lutgendorf MA, Stoll KA (2016) Why 99% may not be as good as you think it is: limitations of screening for rare diseases. J Matern Fetal Neonatal Med 29(7):1187–1189
Maxim LD, Niebo R, Utell MJ (2014) Screening tests: a review with examples. Inhalation Toxicol 26(13):811–828
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J (2011) Scikit-learn: Machine learning in Python. J Mach Learn Res 12:2825–3283
Roh SW, Abell GC, Kim KH, Nam YD, Bae JW (2010) Comparing microarrays and next-generation sequencing technologies for microbial ecology research. Trends Biotechnol 28(6):291–299
Saliba AE, Santos SC, Vogel J (2017) New RNA-seq approaches for the study of bacterial pathogens. Curr Opin Microbiol 35:78–87
SantaLucia J (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci 95(4):1460–1465
Schena M (2003) Microarray analysis. Wiley-Liss
Segawa S, Sawai S, Murata S, Nishimura M, Beppu M, Sogawa K, Watanabe M, Satoh M, Matsutani T, Kobayashi M, Iwadate Y (2014) Direct application of MALDI-TOF mass spectrometry to cerebrospinal fluid for rapid pathogen identification in a patient with bacterial meningitis. Clin Chim Acta 435:59–61
Shulman ST (1997) The biologic and clinical basis of infectious diseases. WB Saunders Company
Šimundić AM (2009) Measures of diagnostic accuracy: basic definitions. Ejifcc 19(4):203
Smith T (1934) Parasitism and disease. Princeton, pp 1–196
Van den Tweel JG, Taylor CR (2010) A brief history of pathology. Virchows Arch 457(1):3–10
Watson DW, Brandly CA (1949) Virulence and pathogenicity. Annu Rev Microbiol 3(1):195–220
Zinsser, H. (1914). Infection and the problem of virulence. In: Infection and resistance. The Macmillan Company, New York, NY, pp 1–27.
Acknowledgements
The use of the HPC facilities at the U of Memphis and the use of GenBank and CDC for publicly available data are gratefully acknowledged. The authors also thank anonymous reviewer(s) for comments and suggestive questions that helped improved the content and presentation of this work.
Funding
The author(s) received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
MG is responsible for the original idea of computational pathogenicity, coordinating the project, the ideas behind the computation of genomic signatures, the PNP tests, preprocessing the downloaded data and computing the Gibbs energies and h-distances. SM downloaded the bacterial and nonpathogenic fungi data, computed genomic signatures, trained, and tested most of the machine learning models on Python software and visualized the results in the figures.
MC helped identify the data, provided background for the introduction and the assessment of the methods and the appropriate metrics from the fields of immunology and pharmacology. SA wrote the custom scripts in MatLab, ran the PNP tests, downloaded sequences for nonpathogenic fungi, trained a few machine learning models and tabulated the results. All authors jointly analyzed the models and they agreed on the conclusions.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval and consent to participate
This research did not require any ethics board approval since no living organisms were involved.
Additional information
Communicated by Shuhua Xu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Garzon, M., Mainali, S., Chacon, M.F. et al. A computational approach to biological pathogenicity. Mol Genet Genomics 297, 1741–1754 (2022). https://doi.org/10.1007/s00438-022-01951-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-022-01951-w