
Abstract
Previous studies revealed important roles of small RNAs (sRNAs) in regulation of bacterial metabolism, stress responses and virulence. However, only a minor fraction of sRNAs is well characterized with respect to the spectra of their targets, conditional expression profiles and actual mechanisms they use to regulate gene expression to control particular biological pathways. To learn more about the specific contribution of sRNAs to the global regulatory network controlling the Escherichia coli central carbon metabolism (CCM), we employed microarray analysis and compared transcriptome profiles of E. coli cells grown on two alternative minimal media supplemented with either pyruvate or glucose, respectively. Microarray analysis revealed that utilization of these alternative carbon sources led to profound differences in gene expression affecting all major gene clusters associated with CCM as well as expression of several known (CyaR, RyhB, GcvB and RyeA) and putative (C0652) sRNAs. To assess the impact of transcriptional reprogramming of gene expression on E. coli protein abundance, we also employed two-dimensional protein gel electrophoresis. Our experimental data made it possible to determine the major pathways for pyruvate assimilation when it is used as a sole carbon source and reveal the impact of other key processes (i.e., energy production, molecular transport and cell resistance to stress) associated with the CCM in E. coli. Moreover, some of these processes were apparently controlled by GcvB, RyhB and CyaR at the post-transcriptional level, thus indicating the complexity and interconnection of the regulatory networks that control CCM in bacteria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pervasiveness of bacteria and their persistence in adverse environments are the results of their evolved ability to adapt and survive under hostile and continuously changing conditions. These properties of bacteria are associated with their capacity to efficiently utilize the resources available in their surroundings and to promptly adjust their metabolism in response to environmental changes. Among the cellular factors that control bacterial fitness and adaptation, trans-encoded small RNAs (sRNAs) are known to play one of the key roles (Kaberdin and Bläsi 2006). Previous work revealed their contribution to regulation of biofilm formation (Bardill et al. 2011), anaerobic growth (Murashko and Lin-Chao 2017), virulence (Porcheron et al. 2014) and chemotaxis (Papenfort et al. 2009). Moreover, a number of sRNAs are involved in responses to environmental cues and stress conditions such as limitation of iron (Masse et al. 2007) and exposure to reactive oxygen species (Altuvia et al. 1997). Some sRNAs (e.g., Spot42, SgrS, GcvB (Wagner and Romby 2015)) also contribute to the control of central carbon metabolism (CCM). For instance, two well-known sRNAs, Spot42 and SgrS, exert their control of CCM by participating in regulation of galactose metabolism and glucose-phosphate stress response, respectively (Møller et al. 2002; Vanderpool and Gottesman 2004).
The Escherichia coli CCM comprises biochemical pathways that are important for generation of energy and production of intermediate metabolites (Noor et al. 2010). The latter serve as precursors for biosynthesis of amino acids and other essential cellular components (Noor et al. 2010). Despite a large number of studies that have contributed to the discovery of the key reactions and metabolites that take part in bacterial carbon metabolism, still the regulatory networks that control the expression of genes involved in CCM are not fully defined, and current studies continue to reveal new features (Basan et al. 2015; Cao et al. 2016; Morin et al. 2016). Moreover, the complexity and interconnection of metabolic pathways suggest that a rewiring of CCM during adaptation to different carbon sources likely involves post-transcriptional mechanisms mediated by known and putative sRNAs.
To gain further insights into the regulatory networks and key factors controlling the CCM in E. coli, we compared gene expression profiles of E. coli cells grown in M9 minimal media supplemented with either pyruvate or glucose (M9/pyruvate and M9/glucose). The use of pyruvate and glucose as alternative carbon sources made it possible to compare the transcriptome profiles of E. coli cells that grow by utilizing either glycolytic (growth on M9/glucose) or gluconeogenetic (growth on M9/pyruvate) pathways. Analysis of gene expression differences was carried out by employing a custom microarray recently developed for simultaneous profiling of protein-coding genes and sRNAs in E. coli (Ruiz-Larrabeiti et al. 2016). Moreover, the results of the transcriptome profiling were further compared with protein expression data obtained by two-dimensional protein gel electrophoresis.
We found that, in addition to the regulation of protein-coding genes whose products directly contribute to CCM, a number of genes encoding several sRNAs (CyaR, RyhB, GcvB, RyeA, GlmY and RprA) with known or predicted roles in E. coli metabolism and stress responses (reviewed in Richards and Vanderpool 2011; Michaux et al. 2014) were differentially expressed as well. Analysis of gene expression patterns associated with E. coli growth on pyruvate versus glucose made it possible to reveal the major routes of pyruvate metabolism, their dependence on other cellular processes and specific roles of small RNAs in E. coli adaptation to different carbon sources.
Materials and methods
Strains and growth conditions
The experiments described here were performed using E. coli K-12 MG1655 strain (Blattner et al. 1997) routinely grown at 37 °C with shaking (200 rpm) in M9 minimal medium supplemented with 0.4% (w/v) glucose (Sigma-Aldrich) or 0.48% (w/v) pyruvate (Sigma-Aldrich). The above concentrations of glucose and pyruvate were chosen to provide equivalent amounts of carbon sources.
RNA isolation
Total RNA was isolated by using a modified hot phenol RNA extraction method (Khodursky et al. 2003; Ruiz-Larrabeiti et al. 2016). Briefly, aliquots of cell cultures grown in triplicate in M9/pyruvate or M9/glucose medium at 37°C with shaking (200 rpm) until OD460 ∼ 0.4–0.6 were mixed with cold stop solution (5% phenol in ethanol) at an 8:1 ratio. The cells were collected by centrifugation (15 min, 4 °C, 4000g), suspended in 250 μL of 1X TE (pH 8.0) supplemented with lysozyme (0.5 mg/ml; Sigma) followed by addition of 1/10 volume of 10% SDS and further incubation at 95 °C for 30 to 60 s. RNA was consequently extracted from the obtained cell lysates by acid phenol (pH 4.5), chloroform and then precipitated with isopropanol. The resulting pellet of RNA was washed with 80% ethanol, air-dried for 15 min and dissolved in RNase-free water. To remove remaining genomic DNA, RNA was further treated with the RNase-free DNase I (Thermo Scientific) for 60 min at 37 °C, extracted by acid phenol (pH 4.5) and chloroform and precipitated with isopropanol. The resulting RNA pellets were washed with 80% ethanol, air-dried and suspended in RNase-free water.
Microarray analysis
Cell cultures were grown in triplicate in M9/glucose or M9/pyruvate medium at 37 °C with shaking (200 rpm) until OD460 ∼ 0.5 and isolation of total RNA including its treatment with DNase I was performed following the protocol described above. After testing the quality and integrity of RNA by using an Agilent 2100 Bioanalyzer, RNA was converted to cDNA and coupled to fluorescent dyes (Cy5 or Cy3) with the SuperScript Indirect cDNA labeling System (Invitrogen), and subsequently used for hybridization with custom microarrays (Ruiz-Larrabeiti et al. 2016) manufactured by Agilent Technologies.
Raw data were acquired from microarray slides with Feature Extraction Software (Agilent Technologies), then further processed by GeneSpring MultiOmic Analysis Software 11.5.1 (Agilent Technologies) and finally subjected to LIMMA statistical analysis by employing WebMeV (Multiple Experiment Viewer) available at http://mev.tm4.org/ (Saeed et al. 2003, 2006). The processed microarray data were deposited in the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) with accession number E-MTAB-5432.
Northern blotting
Northern blotting was performed essentially as described previously (Ruiz-Larrabeiti et al. 2016). Briefly, total RNA was isolated from E. coli K-12 MG1655 strain grown in M9/glucose or M9/pyruvate. Ten micrograms of total RNA were separated by denaturing electrophoresis on 6% polyacrylamide–urea sequencing gels, electro-transferred to a nylon membrane, UV cross-linked, and hybridized with 5′-32P-labeled oligonucleotide probes (see Table S1) complementary to the corresponding transcripts. The size marker (ladder) was produced by hybridization with a mixture of four 5′-32P-labeled probes complementary to tRNA (95 nt), 5S rRNA (120 nt), 6S RNA (183 nt) and RnpB RNA (M1) (377 nt), respectively. Following hybridization at 42 °C overnight, the membranes were washed twice with 20 ml of pre-heated wash buffer [5X SSC with 0.5% (w/v) SDS] at 49 °C for 30 min, then exposed to Imaging Screen-K (Bio-Rad) and the images of the radioactive signals were captured by Molecular Imager FX (Bio-Rad).
Preparation of the total cell lysates
Escherichia coli cells were grown in 500 ml of M9/glucose or M9/pyruvate medium at 37 °C with shaking (200 rpm) until OD460 ∼ 0.5 and then were pelleted by centrifugation (4000g, 10 min, 4 °C). Each pellet was resuspended in 6 ml of TBS buffer (50 mM Tris·HCl, 100 mM NaCl, 15 mM EDTA, pH 8.0) supplemented with 2 mM PMSF and protease inhibitor cocktail (pancreas extract 0.02 mg/ml, thermolysin 0.0005 mg/ml, chymotrypsin 0.002 mg/ml and papain 0.33 mg/ml, Sigma). The cells were disrupted by passing through a MC cell breaker (Multi-shot Cell Disruptor, Pressure BioSciences Inc.) at 39.5 mPa and then cell debris, remaining intact cells and possible protein aggregates were removed by centrifugation at 15,000g (4 °C) for 30 min. The supernatants (S15 fractions) were further filtrated through 0.2 μm pore size filters (Sigma). The protein concentration in each fraction was determined with a Nanodrop 2000 UV Spectrophotometer (Thermo Scientific).
Two-dimensional protein gel electrophoresis
To prepare samples for 2D electrophoresis, aliquots of S15 fractions (each containing equal quantities of total proteins) were individually concentrated by precipitation with trichloroacetic acid (TCA) as described previously (Link and LaBaer 2009). Briefly, aliquots containing ca. 1 mg of soluble proteins were mixed (9:1) with to 100% TCA and incubated on ice for 10 min followed by centrifugation (12,000g, 4 °C, 10 min). The protein pellets were washed twice with 1 ml cold acetone to remove remaining TCA, centrifuged again (13,000g, 5 min, 4 °C) and, after removal of acetone, the pellets were briefly dried at 95 °C for 3 min to evaporate remaining acetone completely. Each pellet was solubilized in 2D rehydration buffer [8 M urea, 2% CHAPS, 2% (v/v) IPG buffer (pH 3–10 NL, GE Healthcare Life Sciences), 0.002% bromophenol blue and 0.2% (w/v) DTT ] at 4 oC for 3–4 h and solutions of solubilized proteins (1 µg/µl) were stored at – 80 °C until further use.
Prior isoelectric focusing (IEF), protein samples were thawed at room temperature, briefly (1–2 min) vortexed and centrifuged for 5 min at 13,000g. Aliquots of supernatants (250 µL each) that contained 250 µg of the solubilized proteins were applied onto 13-cm Immobiline DryStrip gels with immobilized pH gradient (IPG) ranging from 4 to 7 (GE Healthcare Life Sciences). The rehydration was carried out for 20 h, at 120 V followed by IEF (500 V (1 h), 500 V to 1000 V (1 h), 1000 V to 8000 V (3 h), 8,000 V (2 h) in an Ettan IPGphor II Isoelectric Focusing System (Amersham-Pharmacia, Sweden) at 20 °C with 50 μA per strip. After IEF, the strips were equilibrated in equilibration buffer (7 M urea, 2 M thio-urea, 1% DTT, 4% (w/v) CHAPS, 2% IPG buffer (pH 3–10 NL), 0.02% bromophenol blue, 2% DTT) for 12–15 min and then for 6–15 min in the fresh equilibration buffer (without DTT) supplemented with 5% iodoacetamide. As soon as the equilibration was completed, the strip was sealed with melted agarose sealing solution (1X SDS electrophoresis buffer, 1% agarose and 0.02% bromophenol blue) on the top of a 12% SDS polyacrylamide gel and further electrophoretic fractionation of proteins was performed in the second dimension. After electrophoresis, the gels were stained with Coomassie brilliant blue G-250. The images of the stained gels were captured by scanning and were subsequently analyzed by the ImageMaster™ 2D Platinum v7.0 software (GE Healthcare).
Mass-spec identification of proteins
Gel pieces containing the selected protein spots were excised manually from the gels, cut into small pieces and subjected to in-gel digestion according to a modified protocol described previously (Shevchenko et al. 2006). Briefly, the excised gel pieces were washed with the solution containing 25 mM NH4HCO3 in methanol/acetonitrile (2:3 v/v); proteins were reduced with DTT and alkylated with iodoacetamide, and then washed with water and vacuum dried. Further trypsin digestion was performed with trypsin solution (12.5 ng/μL trypsin in 25 mM ammonium bicarbonate containing 10% (v/v) acetonitrile) for 12-16 h at 37°C. The reaction was stopped by adding 5% formic acid. An aliquot (0.5 μL) of the digest was rigorously mixed with 0.5 μL matrix solution (5 mg/ml 2,5-dihydroxybenzoic acid, 0.1% trifluoacetic acid and 30% acetonitrile) and 0.3 μL of the resulting mixture was deposited onto the 384/600-μm MTP AnchorChip (Bruker Daltonik GmbH) chip. Further MALDI-TOF MS mass-spectrometric analysis was performed in positive ion mode with delayed extraction (reflection mode) on a Bruker Autoflex III MALDI TOF/TOF mass spectrometer (Bremen, Germany) equipped with a 200 Hz SmartBean Laser. Data acquisition and processing were done manually by using FlexControl 3.4 and Flex-Analysis 3.4 (Bruker Daltonik GmbH), respectively. The mass spectrometric data were further processed with the Biotools 3.2 (Bruker) package by assessing the online Mascot server (www.matrixscience.com) to identify the corresponding polypeptides and obtain more information about the identified proteins the Swiss-Prot database. For protein identification, the following parameters were set: carboxyamidomethylation on cysteine as a fixed modification, oxidation of methionines as a variable modification, 60 ppm of peptide mass tolerance, 0.7 Da fragment mass tolerance and 2 missed cleavages.
Results
Gene expression analysis
To obtain a global overview of the gene co-expression network involved in coordination of central carbon metabolism and related pathways, we performed transcriptional profiling of E. coli cells grown in M9 minimal media supplemented with either pyruvate or glucose by using a custom microarray previously developed in our group (Ruiz-Larrabeiti et al. 2016). The group of transcripts that showed at least twofold difference in the expression level in cells grown in M9/pyruvate when compared to those grown in M9/glucose were further analysed with the STRING database software (Franceschini et al. 2013) to cluster the differentially expressed genes according to their biological roles. This analysis revealed several groups of up- and downregulated genes involved in CCM and related pathways (Table 1, Fig. 1). The data obtained revealed that during their growth on pyruvate, E. coli cells expressed numerous genes involved in gluconeogenesis (e.g., pck, fbp) and pyruvate-dependent pathways (ackA, acs) more efficiently than when E. coli was grown on glucose. In contrast, the transcripts encoding the enzymes that function in glycolysis (e.g., pykF, eno), sugar storage (e.g., otsA, glgB), pentose phosphate pathway (talA, tktB) and mixed acid fermentation (adhE, adhP, tdcE) were more abundant when glucose was used as an alternative carbon source. Among the differentially expressed genes, there were also those involved in biosynthesis and degradation of cellular components (e.g., fatty acids, amino acids, nucleotides, vitamins and cofactors) essential for regular cell growth (Table 1).

Differentially expressed genes involved in the E. coli central carbon metabolism. Comparisons of transcriptome profiles of E. coli cells grown on pyruvate versus glucose revealed that the level of some CCM-related transcripts was higher (red), lower (blue), or remained nearly the same (black). The main routes of pyruvate assimilation are indicated and numbered (colour figure online)
In addition, microarray analysis also revealed differences in expression level of genes controlling other cellular functions, such as stress response, genetic information processing and electron transport. The full set of genes grouped according to their biological functions is presented in Table S2.
Small RNAs differentially expressed in E. coli cells grown on pyruvate versus glucose
Apart from probes hybridizing with regular protein-coding transcripts, our custom microarray also contained probes specific for known and putative sRNAs (Ruiz-Larrabeiti et al. 2016). Based on analysis of microarray data obtained with sRNA probes, we could identify several known (RyeA, CyaR, RyhB, GcvB, GlmY, RprA) and one putative (C0652, (Carter et al. 2001; Tjaden et al. 2002)) sRNA genes that were differentially regulated in cells grown on pyruvate versus glucose (Table 2). All of them were expressed at higher levels in cells grown on pyruvate.
To validate the expression patterns of these sRNAs, total RNA isolated from cells separately grown on pyruvate and glucose was additionally subjected to Northern blot analysis. Five sRNAs (i.e., RyeB, CyaR, GcvB, RyhB and C0652) that showed greater differences in expression levels according to the Northern blot data (Fig. 2) were selected for further analysis.

Northern blotting of known and putative sRNAs differentially expressed according to microarray data. Detection of sRNAs was performed by using total RNA isolated from E. coli cells grown in M9/glucose or M9/pyruvate (lanes G and P, respectively) and further hybridized to the specific oligonucleotide probes listed in Table S1. Molecular ladder (shown on the right) was separately obtained by hybridization of total RNA with radiolabelled probes (see Table S1) specific for E. coli M1 RNA (377 nt), 6S RNA (183 nt), 5S rRNA (120 nt) and tRNAAsn (75 nt). The equal amounts of total RNA in each separate blot was verified by probing of 5S rRNA, respectively, shown under G and P lanes of each panel
Assessing the effect of alternative carbon sources on protein abundance by two-dimensional gel electrophoresis
To relate the level of transcripts and products of their translation, the abundance of E. coli polypeptides was assessed by 2D electrophoresis of total cell extracts prepared from three biological replicates of E. coli cells grown on M9 supplemented with glucose or pyruvate, respectively. A representative pair of images is shown in Fig. 3. A number of polypeptides (Fig. 3), primarily those with altered abundances, were further identified by mass-spectroscopy and the results of their identification are presented in supplementary Table S3. Consistent with the outcome of the transcriptome analysis, proteome data revealed that E. coli growth on pyruvate versus glucose led to a decrease in the abundance of the key enzymes involved in glycolysis [e.g., GapA (spot 65), TpiA (spot 28, GpmA (spot 29)], TCA cycle [e.g., SucC (spot 61), SucD (spot 21), amino acid (e.g., CysK (spot 65) and TrpB (spot 53)] and nucleoside [e.g., PurH (spot 43) and Upp (spot 25)] metabolism. Moreover, apart from the anticipated changes in concentration of proteins/enzymes involved in CCM, growth on alternative carbon sources differentially affected transcription [RpoA (spot 64)], protein synthesis and turnover [e.g., Frr (spot 73) and DsbA (spot 16)], transport of amino acids [ArgT (spot 23), GltI (spot 19) and GlnH (spot 31)] and cell resistance to oxidative stress [SodB (spot 52)]. The higher levels of the identified proteins usually reflected the higher abundances of their cognate mRNAs detected by microarray analysis (for examples, see the last column of Table S2), thus suggesting that the regulation of the respective genes was primarily exerted at the transcriptional level. In contrast, the relative abundance of 4 polypeptides (i.e., PckA, MaeB, AldA and TnaA; Table S2) was inversely correlated with the abundance of the corresponding mRNAs. Although the specific mechanisms controlling the level of these polypeptides under the conditions tested in this study are largely unknown, they are likely exerted at both transcriptional and post-transcriptional levels.

Two-dimensional analysis of proteins differentially expressed in E. coli cells grown on pyruvate (a) vs glucose (b). Proteins (250 µg) present in total cell lysates were concentrated by precipitation with TCA, subjected to isoelectric focusing and further fractionated in 12% SDS polyacrylamide gels as described in “Materials and methods”. After Coomassie blue staining, the polypeptides differentially expressed in E. coli grown on alternative carbon sources were identified by mass-spectroscopy. The location of the identified polypeptides that were expressed at higher relative levels in cells grown on pyruvate (a) and glucose (b) or showed similar abundance is indicated by red, blue or black circles, respectively. The names of the identified polypeptides are shown
Known and putative sRNA targets differentially expressed in E. coli cells grown on pyruvate vs glucose
Further analysis of microarray data disclosed that higher levels of RyeB, CyaR, GcvB and RyhB in cells grown on pyruvate correlated with a decrease in the abundance of their known target mRNAs and/or their products of translation (for details, see Table S4). In addition, our gene expression data also revealed that a number of putative sRNA targets (see Table S5) envisaged in previous studies (Melamed et al. 2018) had likewise lower abundance in cells grown on pyruvate and therefore were potentially controlled by new antisense mechanisms mediated by RyeB, CyaR, GcvB or RyhB in vivo. Moreover, as the post-transcriptional control of some genes by these sRNAs might affect protein abundance (e.g., regulation of SodB level by RyhB, Table S4), we additionally analyze 2D protein electrophoresis data to search for correlation between the level of the differentially expressed polypeptides and sRNAs predicted to control the corresponding protein-coding genes (Melamed et al. 2018; Chao et al. 2017). This analysis made it possible to expand the number of putative genes and their products (see Table S6) whose level might potentially be controlled by RyeB, CyaR, GcvB, RyhB via new post-transcriptional mechanisms during E. coli growth on alternative carbon sources.
Discussion
A profound understanding of bacterial adaptation to different and often challenging environmental conditions necessarily includes comprehensive insights into gene expression mechanisms controlling CCM and related pathways. Although the key biochemical routes that belong to the bacterial CCM have been extensively studied, the map of regulatory network that control and fine-tune CCM is still under construction and merits further analysis. To obtain a global overview of the gene expression changes adjusting the level of the key enzymes and regulatory factors associated with the E. coli CCM, we compared transcriptome profiles of E. coli cells grown in M9/pyruvate versus M9/glucose. Glucose is the preferred carbon source in E. coli (Inada et al. 1996; Stulke and Hillen 1999; Bruckner and Titgemeyer 2002; Aidelberg et al. 2014) and it is normally metabolized through glycolysis and alternative routes such as Entner–Doudoroff and pentose pathways.
Consistent with the current view of the central carbon metabolism (Holms 1996), substitution of glucose by pyruvate in the growth medium led to (1) a decrease in the relative expression of genes involved in glycolysis and sugar metabolism (i.e., mixed acid fermentation, starch and sucrose metabolism, pentose phosphate and Entner–Doudoroff pathways, sugar storage) and (2) a concomitant increase in the relative expression of genes involved in gluconeogenesis, pyruvate metabolism, acetate assimilation and related pathways (Table 1, Fig. 1). According to the information presented in the KEGG database (https://www.genome.jp/kegg/pathway.html), pyruvate can be metabolized, at least, via eight alternative routes involving different enzymes. Our gene expression analysis, however, revealed that only three genes (ppsA, ldhA and ilvM) encoding enzymes that, respectively, convert pyruvate to phosphoenol pyruvate (PEP), lactate and (S)-2-acetolactate were upregulated, thereby suggesting three primary pathways for pyruvate assimilation (i.e., pathways 1, 5 and 8; Fig. 1). In contrast, expression of other genes involved pyruvate catabolism (i.e., eda, tdcE, poxB, maeB, alaA) via its conversion to 2-dehydro-3-deoxy-d-gluconate 6-phosphate, acetyl-CoA, acetate, (S)-malate and l-alanine was readily reduced.
Further analysis of gene expression data suggested that E. coli seems to reprogram gene expression not only to optimize CCM but also to adjust other physiological functions essential for balanced cell growth. This could be particularly relevant as E. coli grows twofold slower on M9/pyruvate than on M9/glucose (data not shown). We found that the slower growth rate on pyruvate correlated with the overall reduction in the efficiency of biosynthetic pathways, energy production and other essential cellular functions indirectly dependent on the use of alternative carbon sources. In particular, microarray data revealed that E. coli growth on pyruvate led to a decrease in expression of genes involved in biosynthesis of amino acids (i.e., dapB, lysC), nucleosides and nucleotides (i.e., guaA, purB), vitamins and cofactors (i.e., nadB, pntA) and fatty acids (i.e., accB, fabA) as compared to its growth on glucose. In contrast, many catabolic genes involved in control of recycling and degradation of essential cellular components (Fig. 1, Table 1 and S2) including fatty acids (i.e., fadA, fadE), amino acids (i.e., paaJ, tdh), and cell envelope components (i.e., ampD, ldcA) were expressed at higher levels (Fig. 1, Table 1 and S2), thus suggesting that growth rate differentially affects anabolic and catabolic pathways. Similar to expression of biosynthetic genes, a number of genes involved in the processing of genetic information (DNA replication, folding and repair, transcription, translation, RNA folding, ribosome biogenesis, nucleoside and nucleotide metabolism, etc.) also showed lower relative expression in cells grown on pyruvate as compared to glucose. Consistent with the lower replication rate of DNA in cells grown on pyruvate, the cspD gene encoding an inhibitor of DNA replication was highly expressed.
Besides its effects on the expression of metabolic genes, the growth-rate-dependent control was apparently in charge of the differential expression of nearly all genes related to generation of energy (see Table S2). This process was less efficient in cells grown on pyruvate and therefore could limit other physiological functions such as biofilm formation and motility. Indeed, the expression of the corresponding genes (e.g., dosC, ydaM and fliO, fliN and fliM) was reduced during E. coli growth on pyruvate and resembled their growth-rate-dependent regulation observed in other studies (Sim et al. 2017).
In addition, we found that E. coli growth on different carbon sources altered the expression of genes involved in bacterial adaptation to stress. The majority of them encoded enzymes with known roles in resistance to oxidative stress (e.g., katE, katG, ychH, sodB and marRAB) and acid stress (i.e., gadE, gadX and hdeD). Moreover, control of some stressed-related genes (e.g., sodB, marA, hdeD) was apparently exerted by sRNAs at the post-transcriptional level (see below). The observed downregulation of some genes that are involved in oxidative stress response during E. coli growth on pyruvate versus glucose was likely linked to the higher growth rate characterized by more abundant energy production through oxidative phosphorylation raising the level of reactive oxygen species (ROS). Since ROS are normally toxic for cells, bacteria need to cope with the oxidative stress by upregulating the corresponding genes.
Another large group of the differentially expressed genes was related to molecular transport functions. Most of the transcripts encoding trans-membrane importers involved in the uptake of cellular components such as nucleotides (i.e., uraA, codB), amino acids (i.e., gadC, mmuP) and compounds containing metallic ions (i.e., btuD, fhuB) as well as exporters represented by bacterial secretion systems and drug-proton antiporters (i.e., emrA, gspC) were less abundant in cells grown on pyruvate. In contrast, only few transporter-encoding genes were upregulated. The most prominent ones included two genes coding for the BtsSR pyruvate sensor system (i.e., btsS and btsR) as well as major pyruvate transporter genes (i.e., btsT, yhjX and cstA (Hwang et al. 2018).
Increases in expression were also observed for the phoS, pstA and pstC genes encoding the components of the ATP-dependent phosphate ABC transporter. The anticipated increase in the uptake of phosphate required for phosphoenol pyruvate (PEP) formation along with the concomitant upregulation of the ppsA gene coding for PEP syntase suggest that conversion of pyruvate to PEP might represent the key pathway for pyruvate assimilation when it is used as a sole carbon source. Interestingly, the upregulation of ZnuA, a high-affinity transporter involved in Zn2+ uptake, can have an indirect implication in the control of CCM. The considerably higher level of this transporter in cells grown on pyruvate (see Fig. 3, spot #13) apparently increases the level of zinc ions. The latter have inhibiting effects on the key enzymes of glycolysis [PGM, PFK and GAPDH; (Ong et al. 2015)] and therefore might play important regulatory roles in reprogramming the E. coli CCM. Besides its inhibitory effect on glycolysis, an increase in Zn uptake could likely influence the activity of Zn-dependent enzymes such as carbonic anhydrase (Merlin et al. 2003) potentially important for E. coli growth on pyruvate. In contrast to upregulation of zhuA, a number of genes involved in uptake of silver, copper and iron were downregulated (see Table S2), thus suggesting a decrease in the use of these metal ions in E. coli cells grown on pyruvate.
In addition, our microarray data revealed differential regulation of several known and putative sRNAs (Table 2). Moreover, the level of some well-known sRNAs (i.e., GcvB, CyaR and RyhB) was considerably higher in cells grown on pyruvate. The first two sRNAs (i.e., CyaR and GcvB) are primarily known for their contribution to regulation of CCM (Papenfort and Vogel 2009), whereas RyhB is widely recognized for its role in regulation of iron homeostasis and oxidative stress response. Although there are more E. coli sRNAs (i.e., Spot 42, SgrS, and CsrB, CsrC) that are also known for their contribution to regulation of CCM (see Introduction), we did not observe any significant changes in their levels. The latter may suggest that the specific metabolic pathways (i.e., galactose metabolism, sugar-phosphate stress or carbon storage, respectively) that are controlled by these sRNAs were likely not affected to a significant degree upon switching from glucose to pyruvate catabolism.
GcvB was demonstrated to regulate a number of CCM-related genes (reviewed in Papenfort and Vogel 2009). We found that a higher level of this sRNA in cells grown on pyruvate was in agreement with the lower abundance of several GcvB targets (e.g., ilvC, livJ, yeaC, ndk, ybdH and ilvE), thus suggesting the GcvB-dependent downregulation of amino acid synthesis (ilvC) and transport (livJ), nucleotide synthesis (ndk) and still unknown functions (ybdH). Furthermore, the negative control exerted on some targets (maeB, dppA, oppA,and argT) was revealed at the protein level (Fig. 3; protein spots 36, 44 (45 and 46) and 47, respectively). However, since few GcvB targets (e.g., gltI and cycA) still remained unaffected, our data suggest that some regulatory functions of GcvB could partly be impaired and this effect was likely dependent on the growth media. Namely, several studies (Urbanowski et al. 2000; Pulvermacher et al. 2009) have recently shown that GcvB is poorly expressed in minimal media and its abundance might not be high enough to overcome the threshold level for interaction with some targets.
Similar to GcvB, CyaR was more abundant in cells grown on pyruvate (Table 2, Fig. 2). Among its known targets, only hdeD (Lalaouna et al. 2018) was found among the genes differentially expressed at the transcriptional level (see Table S2). As the product of the hdeD gene is implicated in acid resistance (Masuda and Church 2003), the reduced expression of this gene could be a result of less efficient acidification conferred by E. coli growth on pyruvate.
Unlike GcvB and CyaR, RyhB is better known for its control of iron homeostasis and iron-dependent enzymes. An increase in RyhB level is normally attributed to relieve of Fur-dependent ryhB repression that occurs under iron-limiting conditions characterized by less efficient production of reactive oxygen species (ROS), thus making some ROS-detoxifying enzymes (e.g., superoxide dismutase B, SodB) redundant. Consistently, we observed a decreased production of SodB (Fig. 3, spot 52) matching the RyhB-mediated downregulation of its cognate transcript (i.e., sodB) in cells grown on pyruvate. Another regulatory function of RyhB is likely linked to a decrease in metabolic and respiratory activities observed during E. coli growth on pyruvate (see the third paragraph of this section). Namely, our data indicate that RyhB could likely inhibit acnB translation, thereby reducing the level aconitase B (AcnB; Fig. 3, spot 34) playing an essential role in the TCA cycle. Interestingly, the decrease in the abundance of iron-dependent enzymes (i.e., SodB and AcnB) was concomitant with downregulation of trxB and grxB encoding thioredoxin reductase TrxB (spot 68) and thioredoxin 4, respectively, involved in iron-sulfur cluster biogenesis (Ding et al. 2005). Moreover, the downregulation of grxB was likely dependent on RyhB (see Lalaouna et al. 2015). These findings disclosed an important role of RyhB in the coordinated response reducing the level of metabolic enzymes and essential co-factors.
The function of RyeA in CCM is less clear. It has recently been shown (Choi et al. 2018) that this sRNA along with SdsR (also known as RyeB) appears to represent a new type of toxin-antitoxin (TA) system. An increase in RyeA level can therefore reduce the negative impact SdsR (toxin) on expression of genes more important for E. coli growth on pyruvate.
In addition to regulation of known sRNAs, microarray analysis also disclosed differential expression of C0652 (see Table 2), a putative sRNA whose existence was predicted in silico (Carter et al. 2001) and later confirmed by microarray analysis (Tjaden et al. 2002). As the mechanism of action and putative targets of C0652 are currently unknown, future work will be necessary to clarify its regulatory functions and impact on CCM.
Lastly, the concomitant changes in the level of GcvB, CyaR, RyhB and their putative targets (Tables S5 and S6) involved in amino acid biosyntheses and sugar metabolism suggest much broader roles of these sRNAs in regulation of E. coli metabolism. Future studies will make it possible to address the direct and indirect roles of GcvB and CyaR, RyhB in regulation of their putative targets (Table S5 and S6) and therefore will provide new insights into the contribution of GcvB, CyaR and RyhB to the control of E. coli CCM at the post-transcriptional level.
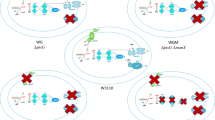
In summary, our study disclosed the complexity of the gene expression network E. coli uses to adjust cell metabolism during bacterial cell growth on pyruvate versus glucose. In particular, we showed that the use of these alternative carbon sources is not merely associated with the multi-level regulation of glycolytic and gluconeogenetic pathways, but is also dependent on mechanisms controlling energy production, molecular transport and cell resistance to stress (Fig. 4a). The study also reveals the major pathways of pyruvate metabolism when it is used as a sole carbon source and suggests that pyruvate conversion to PEP might represent the principal route for pyruvate assimilation. Furthermore, our data suggest that the fine tuning of CCM during E. coli growth on pyruvate versus glucose involves the action of several sRNAs regulating metabolism, transport and adaptation to stress (Fig. 4b). Finally, it is conceivable that metabolic differences associated with the use of these alternative carbon sources along with the possibility to reverse flux from pyruvate to phosphoenolpyruvate (Long et al. 2017) might have important implications in metabolic engineering.

Effect of alternative carbon sources (pyruvate versus glucose) on gene expression in E. coli. a Major biological functions differentially affected in E. coli during its growth on pyruvate versus glucose. b Control of gene expression by small RNAs that are more abundant in cells grown on pyruvate
Change history
20 August 2019
The article Reprogramming of
References
Afonyushkin T, Večerek B, Moll I, Bläsi U, Kaberdin VR (2005) Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res 33(5):1678–1689
Aidelberg G, Towbin BD, Rothschild D, Dekel E, Bren A, Alon U (2014) Hierarchy of non-glucose sugars in Escherichia coli. BMC Syst Biol 8:133. https://doi.org/10.1186/s12918-014-0133-z
Altuvia S, Weinstein-Fischer D, Zhang A, Postow L, Storz G (1997) A small, stable RNA induced by oxidative stress: role as a pleiotropic regulator and antimutator. Cell 90(1):43–53
Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S (2001) Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11(12):941–950
Bardill JP, Zhao X, Hammer BK (2011) The Vibrio cholerae quorum sensing response is mediated by Hfq-dependent sRNA/mRNA base pairing interactions. Mol Microbiol 80(5):1381–1394. https://doi.org/10.1111/j.1365-2958.2011.07655.x
Basan M, Hui S, Okano H, Zhang Z, Shen Y, Williamson JR, Hwa T (2015) Overflow metabolism in Escherichia coli results from efficient proteome allocation. Nature 528(7580):99–104. https://doi.org/10.1038/nature15765
Blattner FR, Gr Plunkett, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277(5331):1453–1462
Bruckner R, Titgemeyer F (2002) Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol Lett 209(2):141–148
Cao Y, Forstner KU, Vogel J, Smith CJ (2016) Cis-encoded small RNAs, a conserved mechanism for repression of polysaccharide utilization in bacteroides. J Bacteriol 198(18):2410–2418. https://doi.org/10.1128/JB.00381-16
Carter RJ, Dubchak I, Holbrook SR (2001) A computational approach to identify genes for functional RNAs in genomic sequences. Nucleic Acids Res 29(19):3928–3938
Chao Y, Li L, Girodat D, Forstner KU, Said N, Corcoran C, Smiga M, Papenfort K, Reinhardt R, Wieden HJ, Luisi BF, Vogel J (2017) In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol Cell 65(1):39–51. https://doi.org/10.1016/j.molcel.2016.11.002
Choi JS, Kim W, Suk S, Park H, Bak G, Yoon J, Lee Y (2018) The small RNA, SdsR, acts as a novel type of toxin in Escherichia coli. RNA Biol 15(10):1319–1335. https://doi.org/10.1080/15476286.2018.1532252
De Lay N, Gottesman S (2009) The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol 191(2):461–476. https://doi.org/10.1128/JB.01157-08
Ding H, Harrison K, Lu J (2005) Thioredoxin reductase system mediates iron binding in IscA and iron delivery for the iron-sulfur cluster assembly in IscU. J Biol Chem 280(34):30432–30437. https://doi.org/10.1074/jbc.M504638200
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41(Database issue):D808–D815. https://doi.org/10.1093/nar/gks1094
Gutierrez A, Laureti L, Crussard S, Abida H, Rodriguez-Rojas A, Blazquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, Matic I (2013) Beta-lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun 4:1610. https://doi.org/10.1038/ncomms2607
Holms H (1996) Flux analysis and control of the central metabolic pathways in Escherichia coli. FEMS Microbiol Rev 19(2):85–116
Hwang S, Choe D, Yoo M, Cho S, Kim SC, Cho S, Cho BK (2018) Peptide transporter CstA imports pyruvate in Escherichia coli K-12. J Bacteriol. https://doi.org/10.1128/jb.00771-17
Inada T, Kimata K, Aiba H (1996) Mechanism responsible for glucose-lactose diauxie in Escherichia coli: challenge to the cAMP model. Genes Cells 1(3):293–301
Johansen J, Eriksen M, Kallipolitis B, Valentin-Hansen P (2008) Down-regulation of outer membrane proteins by noncoding RNAs: unraveling the cAMP-CRP- and sigmaE-dependent CyaR-ompX regulatory case. J Mol Biol 383(1):1–9. https://doi.org/10.1016/j.jmb.2008.06.058
Jorgensen MG, Thomason MK, Havelund J, Valentin-Hansen P, Storz G (2013) Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev 27(10):1132–1145. https://doi.org/10.1101/gad.214734.113
Kaberdin VR, Bläsi U (2006) Translation initiation and the fate of bacterial mRNAs. FEMS Microbiol Rev 30:967–979
Khodursky AB, Bernstein JA, Peter BJ, Rhodius V, Wendisch VF, Zimmer DP (2003) Escherichia coli spotted double-strand DNA microarrays: RNA extraction, labeling, hybridization, quality control, and data management. Methods Mol Biol 224:61–78
Lalaouna D, Carrier MC, Semsey S, Brouard JS, Wang J, Wade JT, Masse E (2015) A 3’ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol Cell 58(3):393–405. https://doi.org/10.1016/j.molcel.2015.03.013
Lalaouna D, Prevost K, Laliberte G, Houe V, Masse E (2018) Contrasting silencing mechanisms of the same target mRNA by two regulatory RNAs in Escherichia coli. Nucleic Acids Res 46(5):2600–2612. https://doi.org/10.1093/nar/gkx1287
Link AJ, LaBaer J (2009) Proteomics: a Cold Spring Harbor Laboratory course manual. CSHL Press, Cold Spring Harbor
Long CP, Au J, Sandoval NR, Gebreselassie NA, Antoniewicz MR (2017) Enzyme I facilitates reverse flux from pyruvate to phosphoenolpyruvate in Escherichia coli. Nat Commun 8:14316. https://doi.org/10.1038/ncomms14316
Majdalani N, Hernandez D, Gottesman S (2002) Regulation and mode of action of the second small RNA activator of RpoS translation, RprA. Mol Microbiol 46(3):813–826
Masse E, Salvail H, Desnoyers G, Arguin M (2007) Small RNAs controlling iron metabolism. Curr Opin Microbiol 10(2):140–145. https://doi.org/10.1016/j.mib.2007.03.013
Massé E, Escorcia FE, Gottesman S (2003) Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17(19):2374–2383
Masuda N, Church GM (2003) Regulatory network of acid resistance genes in Escherichia coli. Mol Microbiol 48(3):699–712
Melamed S, Faigenbaum-Romm R, Peer A, Reiss N, Shechter O, Bar A, Altuvia Y, Argaman L, Margalit H (2018) Mapping the small RNA interactome in bacteria using RIL-seq. Nat Protoc 13(1):1–33. https://doi.org/10.1038/nprot.2017.115
Merlin C, Masters M, McAteer S, Coulson A (2003) Why is carbonic anhydrase essential to Escherichia coli? J Bacteriol 185(21):6415–6424
Michaux C, Verneuil N, Hartke A, Giard JC (2014) Physiological roles of small RNA molecules. Microbiology 160(Pt 6):1007–1019. https://doi.org/10.1099/mic.0.076208-0
Møller T, Franch T, Udesen C, Gerdes K, Valentin-Hansen P (2002) Spot 42 RNA mediates discoordinate expression of the E. coli galactose operon. Genes Dev 16(13):1696–1706
Morin M, Ropers D, Letisse F, Laguerre S, Portais JC, Cocaign-Bousquet M, Enjalbert B (2016) The post-transcriptional regulatory system CSR controls the balance of metabolic pools in upper glycolysis of Escherichia coli. Mol Microbiol 100(4):686–700. https://doi.org/10.1111/mmi.13343
Murashko ON, Lin-Chao S (2017) Escherichia coli responds to environmental changes using enolasic degradosomes and stabilized DicF sRNA to alter cellular morphology. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1703731114
Noor E, Eden E, Milo R, Alon U (2010) Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol Cell 39(5):809–820. https://doi.org/10.1016/j.molcel.2010.08.031
Ong CL, Walker MJ, McEwan AG (2015) Zinc disrupts central carbon metabolism and capsule biosynthesis in Streptococcus pyogenes. Sci Rep 5:10799. https://doi.org/10.1038/srep10799
Papenfort K, Vogel J (2009) Multiple target regulation by small noncoding RNAs rewires gene expression at the post-transcriptional level. Res Microbiol 160(4):278–287. https://doi.org/10.1016/j.resmic.2009.03.004
Papenfort K, Said N, Welsink T, Lucchini S, Hinton JC, Vogel J (2009) Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol Microbiol 74(1):139–158. https://doi.org/10.1111/j.1365-2958.2009.06857.x
Porcheron G, Habib R, Houle S, Caza M, Lepine F, Daigle F, Masse E, Dozois CM (2014) The small RNA RyhB contributes to siderophore production and virulence of uropathogenic Escherichia coli. Infect Immun 82(12):5056–5068. https://doi.org/10.1128/IAI.02287-14
Pulvermacher SC, Stauffer LT, Stauffer GV (2009) The small RNA GcvB regulates sstT mRNA expression in Escherichia coli. J Bacteriol 191(1):238–248. https://doi.org/10.1128/JB.00915-08
Reichenbach B, Gopel Y, Gorke B (2009) Dual control by perfectly overlapping sigma 54- and sigma 70-promoters adjusts small RNA GlmY expression to different environmental signals. Mol Microbiol 74(5):1054–1070. https://doi.org/10.1111/j.1365-2958.2009.06918.x
Richards GR, Vanderpool CK (2011) Molecular call and response: the physiology of bacterial small RNAs. Biochim Biophys Acta 1809(10):525–531. https://doi.org/10.1016/j.bbagrm.2011.07.013
Ruiz-Larrabeiti O, Plagaro AH, Gracia C, Sevillano E, Gallego L, Hajnsdorf E, Kaberdin VR (2016) A new custom microarray for sRNA profiling in Escherichia coli. FEMS Microbiol Lett. https://doi.org/10.1093/femsle/fnw131
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34(2):374–378
Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J (2006) TM4 microarray software suite. Methods Enzymol 411:134–193. https://doi.org/10.1016/S0076-6879(06)11009-5
Sharma CM, Darfeuille F, Plantinga TH, Vogel J (2007) A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev 21(21):2804–2817. https://doi.org/10.1101/gad.447207
Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1(6):2856–2860. https://doi.org/10.1038/nprot.2006.468
Sim M, Koirala S, Picton D, Strahl H, Hoskisson PA, Rao CV, Gillespie CS, Aldridge PD (2017) Growth rate control of flagellar assembly in Escherichia coli strain RP437. Sci Rep 7:41189
Stulke J, Hillen W (1999) Carbon catabolite repression in bacteria. Curr Opin Microbiol 2(2):195–201. https://doi.org/10.1016/S1369-5274(99)80034-4
Tjaden B, Saxena RM, Stolyar S, Haynor DR, Kolker E, Rosenow C (2002) Transcriptome analysis of Escherichia coli using high-density oligonucleotide probe arrays. Nucleic Acids Res 30(17):3732–3738
Urban JH, Vogel J (2008) Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol 6(3):e64. https://doi.org/10.1371/journal.pbio.0060064
Urbanowski ML, Stauffer LT, Stauffer GV (2000) The gcvB gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol Microbiol 37(4):856–868
Vanderpool CK, Gottesman S (2004) Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol Microbiol 54(4):1076–1089
Wagner E, Romby P (2015) Chapter Three—small RNAs in Bacteria and Archaea: who they are, what they do, and how they do it. Adv Genet 90:133–208
Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S (2001) Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 15(13):1637–1651
Acknowledgements
We are grateful to the Gene Expression Unit at the Advanced Core Research Facilities (SGIKer) of the University of the Basque Country (UPV/EHU) for the technical support and assistance provided. We also thank Cheng-Hsilin Hsieh (Genomics Core at the Institute of Molecular Biology) for technical assistance in MALDI-TOF/TOF mass spectrometry.
Funding
This work was in part supported by the Spanish Ministry of Economy and Competitiveness grants BFU2011-25455 and CGL2015-70929-R, Ikerbasque (Basque Foundation for Science), grants AS 034006 and MOST 104-2311-B-001-011-MY3 (to S. L.-C.), a pre-doctoral grant from the University of the Basque Country (O.R-L) and Postdoctoral Fellows Program 23-13 of Academia Sinica (A.C.K.)
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised due to a retrospective Open Access order.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kaberdina, A.C., Ruiz-Larrabeiti, O., Lin-Chao, S. et al. Reprogramming of gene expression in Escherichia coli cultured on pyruvate versus glucose. Mol Genet Genomics 294, 1359–1371 (2019). https://doi.org/10.1007/s00438-019-01597-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01597-1