
Abstract
Bats are hosts for diverse Trypanosoma species, including trypanosomes of the Trypanosoma cruzi clade. This clade is believed to have originated in Africa and diversified in many lineages worldwide. In several geographical areas, including Cameroon, no data about trypanosomes of bats has been collected yet. In this study, we investigated the diversity and phylogenetic relationships of trypanosomes of different bat species in the central region of Cameroon. Trypanosome infections were detected in six bat species of four bat families, namely Hipposideridae, Pteropodidae, Rhinolophidae, and Vespertilionidae, with an overall prevalence of 29% and the highest infection rate in hipposiderid bat species. All trypanosomes were identified as belonging to the Trypanosoma livingstonei species group with one clade that might represent an additional subspecies of T. livingstonei. Understanding the prevalence, distribution, and host range of parasites of this group contributes to our overall knowledge of the diversity and host specificity of trypanosome species that phylogenetically group at the base of the T. cruzi clade.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bats are hosts to a diversity of eukaryotic protozoan parasites, including trypanosomes, Babesia, haemosporidians, and Leishmania (e.g., Gardner and Molyneux 1987; Lima et al. 2013; Schaer et al. 2013; de Souza et al. 2023). Trypanosomes (genus Trypanosoma) are flagellated kinetoplastid blood parasites that are transmitted by leeches and various bloodsucking arthropods and have adapted to infect various classes of vertebrates that comprise several mammalian groups and include species that are a threat to human and animal health (Simpson et al. 2006; Morrison et al. 2016; Büscher et al. 2017). Bats are recognized as hosts for diverse Trypanosoma species, and studies have revealed that the majority of identified bat trypanosomes fall within the Trypanosoma cruzi clade with evidence that bats played an important role in the evolution of the T. cruzi species group (e.g., Austen and Barbosa 2021). However, the knowledge about the diversity of bat trypanosomes, their vectors, distribution, and the evolutionary history of trypanosomes is still limited (e.g., Hamilton et al. 2012; Lima et al. 2013; Clement et al. 2020). In several geographical areas, including Cameroon, no data about trypanosomes of bats has been collected yet. With 112 species of bats, Cameroon is one of the hot spots of bat diversity in Africa (ACR 2022). In the current study, we investigate the diversity and phylogenetic relationships of trypanosomes of different bat species in the central region of Cameroon using molecular methods.
Material and methods
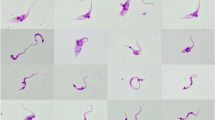
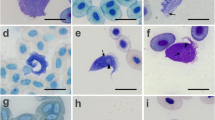
Sampling of bats was conducted in the central region of Cameroon in the dry and wet season between February 2016 and December 2019 across different habitat types like forest, savanna, and cultured farmland as described in Tsague et al. (2022). Bat individuals were captured using ground-level mist nets, and different identification keys were used for morphological species identification (e.g., Rosevear 1965; Patterson and Webala 2012). Small blood samples were collected by venipuncture of the uropatagial vein. A thin blood smear and blood dots on Whatman filter paper (GE Healthcare) were collected from every individual, before it got released at the capture site. The blood smears were dried and fixed in 99–100% (vol/vol) methanol solution for 3 s and subsequently stained with 10% Giemsa solution for 20 min and air-dried. Blood smears were screened for the presence of trypanosome parasites with light microscopy (Leica DMLB 1000) at a magnification of × 400 and × 1000.
The QIAGEN DNeasy blood and tissue extraction kit (Hilden, Germany) was used to extract whole genomic DNA from the dried blood dots following Schaer et al. (2013). PCRs were performed using the AllTaq Master Mix Kit (QIAGEN) with 4 µl DNA and 1 µl of each primer (10 mM). A nested-PCR approach was used for the amplification of about 600 bp of the trypanosome’s small subunit 18S ribosomal RNA gene (18S rRNA) following Noyes et al. (1999) using the primers TRY927F/R for the outer reaction and SSU561F/R for the nested reaction. PCR products were sequenced with the amplification primers and Sanger-sequenced. All nucleotide sequences were assessed for quality and manually edited in the software Geneious Prime 2023.1.2 (https://www.geneious.com) and amplification and sequencing were repeated for samples with low-quality sequences. Trypanosome nucleotide sequences were aligned with reference sequences, obtained from NCBI GenBank, using the MAFFT algorithm (Katoh et al. 2002). The GenBank accession numbers are provided in the respective phylogenetic tree figure. The sequence alignment for the analysis of Trypanosoma taxa comprised 94 sequences and a length of 667 nt. The software modeltest-ng 0.1.7 was used to test different DNA substitution models and the maximum likelihood (ML) analysis was carried out in raxmlGUI 2.0.10 (Darriba et al. 2020) using the model TIM3 + I + G with 10,000 bootstrap iterations and the taxon Trypanosoma lewisi as outgroup. The phylogenetic tree was displayed in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). Haplotype networks were constructed in PopART v.1.7 (Leigh and Bryant 2015) using the median-joining algorithm with default settings. The networks were labeled according to the bat host species from which the Trypanosoma sequence was amplified. Some of the (high-quality) sequences contained ambiguous base calls (with double nucleotide peaks) that point to mixed haplotype infections (Table S1) and were therefore excluded from haplotype network analyses (Table S1).
Results and discussion
A total of 159 bats belonging to four bat families, nine genera, and 13 species were investigated and screened with PCR. Trypanosoma parasite DNA was detected in samples of 46 bat individuals, corresponding to an overall prevalence of 29% (Table 1). Infections were detected in bat species of all four investigated bat families Hipposideridae, Pteropodidae, Rhinolophidae, and Vespertilionidae.
Highest prevalences of trypanosome infections were identified in three bat species of the bat family Hipposideridae, with 76% (19/25) infected individuals of Hipposideros ruber, 70% (7/10) of Doryrhina cyclops, and 38% (13/34) of Hipposideros curtus. No infections were recorded for Hipposideros abae (0/2) and H. fuliginosus (0/19). A low prevalence of trypanosome infections was documented from fruit bats (Pteropodidae), with 9% (3/33) infections in Epomophorus pusillus and none in Eidolon helvum (0/1), Epomops franqueti (0/15), and Rousettus aegyptiacus (0/1), though the sample size of the three latter species was quite low. Two species of Rhinolophus (Rhinolophidae) and two vespertilionid bat species were investigated in the study, with infections verified in one rhinolophid species, Rhinolophus cf. landeri (27%, 3/11) and in Afronycteris nana (20%, 1/5) (Vespertilionidae) (Table 1).
All infections were identified via PCR and sequencing. Comparison of the 18S rRNA trypanosome sequences of the 46 samples with reference sequences on NCBI GenBank (BLASTn) featured highest identity either with sequences of the species Trypanosoma livingstonei/Trypanosoma cf. livingstonei or a closely related Trypanosoma sp. taxon reported from African and European Miniopterus species (Clement et al. 2020; Szentivanyi et al. 2020). Our dataset comprised samples with trypanosome sequences of lower quality, but comparison of these sequences with reference sequences on NCBI GenBank (BLASTn) also featured highest identities with T. cf. livingstonei/livingstonei/sp. parasites (Table S1). High-quality trypanosome sequences from 28 bat samples were included in the subsequent haplotype network and phylogenetic analyses.
The 18S rRNA maximum likelihood phylogenetic analysis recovered the trypanosome sequences of the study within the wider T. livingstonei clade (with high support, bootstrap value of 95) (Fig. 1).

Maximum likelihood (ML) phylogeny of Trypanosoma parasites of the Trypanosoma cruzi clade. The 18S rRNA sequence alignment (length of 667 nt) for the analysis comprised 94 sequences. The ML analysis was carried out using the model TIM3 + I + G with 10,000 bootstrap iterations and the taxon Trypanosoma lewisi as outgroup. Bootstrap values (> 20) are given. All trypanosome sequences of the study (highlighted in bold) fall within the Trypanosoma livingstonei species group (collapsed on the left, uncollapsed on the right). Figure was assembled with BioRender.com
Within this T. livingstonei clade, the Cameroonian bat trypanosome sequences fall in three main subclades. The first main subclade that includes trypanosome sequences of A. nana, E. pusillus, H. ruber, and R. landeri from Cameroon (Fig. 1, highlighted in orange) falls within the group of T. cf. livingstonei parasites that are nested within T. livingstonei parasites. The trypanosome sequences of five H. ruber, A. nana (n = 1), and R. landeri (n = 1) represent one haplotype (haplotype H1), while each sequence of the three E. pusillus bats represents its own haplotype that differs from haplotype H1 by one base each (Supplementary Fig. S1A). Several branches within the T. livingstonei/T. cf. livingstonei are not well supported and a more complete taxon sampling and additional molecular markers are necessary to resolve the relationships among the wider T. livingstonei parasite clade (Fig. 1).
The second subclade includes the aforementioned Trypanosoma sp. clade from Miniopterus bat species (here termed Trypanosoma sp. A) and two sequences from H. curtus from Cameroon (Fig. 1, highlighted in blue) that group as sister group to Trypanosoma sp. A (with low support, bootstrap value = 68). Basal to this group, trypanosome sequences from R. landeri and D. cyclops (Fig. 1, highlighted in blue) from Cameroon form a separate clade albeit with low support (bootstrap value = 49). The sequences from H. curtus represent three different haplotypes, while the sequences of R. landeri and D. cyclops (n = 2) share one haplotype (Supplementary Fig. S1B). The third subclade, here termed Trypanosoma sp. B and supported with a high bootstrap value of 94, comprises two trypanosome parasites from Nigerian bat hosts plus the trypanosome sequences from H. ruber and the remaining sequences from H. curtus from Cameroon (Fig. 1, highlighted in red). Both main subclades, the wider Trypanosoma sp. A and B clades, group as sister clades with high support (bootstrap value 92). So, despite bat host specific clustering of some sequences (e.g., some trypanosome sequences of H. ruber and H. curtus), the results of shared haplotypes and phylogenetic clades of trypanosomes from different bat species point to an overall low host species specificity.
Unfortunately, no trypanosome parasite stages were detected in any of the blood smears of the infected bat samples which points to subpatent/low parasitemia trypanosome infections. In many wildlife hosts, trypanosome infections can be chronic and asymptomatic, leading to low levels of parasites in the bloodstream (e.g., Njiokou et al. 2006). Therefore, the research of wildlife trypanosomes often involves the use of hemoculture to culture trypanosomes from blood samples. This method helps in isolating and identifying different trypanosome species, facilitating, e.g., the microscopic study of the parasite morphology and provides large amounts of DNA for molecular and phylogenetic analyses (e.g., Lima et al. 2013). However, for our current study, we did not collect sufficient amounts of blood from each bat that would have been required for hemoculture. Thus, the findings of the study present a first snapshot of the diversity and prevalence of trypanosome taxa in bats in Central Cameroon, but further studies that include morphological characterization of the parasites and facilitate the analysis of additional phylogenetic markers are needed.
The trypanosome species T. livingstonei was originally described in bats from Mozambique (Lima et al. 2013). Since then, T. livingstonei, its putative subspecies T. cf. livingstonei, and the closely related Trypanosoma sp. A (Clement et al. 2020; Szentivanyi et al. 2020) have been reported from a diversity of African bat species, including the six different bat species in this study (e.g., Clement et al. 2020; Kamani et al. 2022; Thiombiano et al. 2023). The results recovered another subclade of T. livingstonei, the trypanosomes of the Trypanosoma sp. B group. Our data confirm and enlarge the diversity of the T. livingstonei species group, especially among trypanosomes of African bat species. Understanding the prevalence, distribution, and host range of parasites of the T. livingstonei parasite group contributes to our overall knowledge of the diversity and host specificity of trypanosomes species that originated from Africa and phylogenetically group at the base of the T. cruzi clade (Clement et al. 2020; Austen and Barbosa 2021). The species T. cruzi causes Chagas disease in humans and therefore identification and research of closely related trypanosome species is of importance (e.g., Beltz 2017). Understanding the diversity and phylogenetic relationships of bat trypanosomes is crucial for improving our knowledge of the broader group of parasites (Hamilton et al. 2012; Lima et al. 2012). Numerous trypanosome lineages within the T. cruzi clade may have originated in African bat species (e.g., Lima et al. 2013; Clement et al. 2020), highlighting the importance of targeted systematic sampling and molecular characterization of trypanosome species from African bats.
Of note, for the two bat species D. cyclops and E. pusillus, a high incidence of co-infections of trypanosomes and haemosporidian parasites was discovered. The haemosporidian infections in the samples of this study were identified in a previous study that used the same samples and, at that time, focused exclusively on infections with haemosporidian parasites (Tsague et al. 2022). Six out of the seven trypanosome-infected D. cyclops individuals featured infections with Nycteria parasites, while the three trypanosome-infected E. labiatus individuals were co-infected with Hepatocystis parasites (Tsague et al. 2022). To the best knowledge of the authors, this is the first time that co-infections with the two unrelated eukaryotic blood parasites, trypanosomes and haemosporidians (Hepatocystis or Nycteria), have been documented. The co-infections could be a result of a common transmission mechanism or a shared arthropod vector. However, the vectors for the trypanosomes of the study are unknown as are the vectors for Nycteria parasites (e.g., Schaer et al. 2015). Bat Hepatocystis parasites might be vectored by species of Culicoides (Ceratopogonidae) as has been shown for the monkey-infecting Hepatocystis species, H. kochi (Garnham et al. 1961). Further research is needed to explore whether the co-infections of the two different eukaryotic blood parasites are a common phenomenon in some (African) bat host species.
Data availability
All trypanosome sequences of the study are available at GenBank (NCBI) with the accession numbers PP320471–PP320496.
References
ACR (2022) African Chiroptera report 2022. In: Van Cakenberghe V, Seamark ECJ (eds) AfricanBats NPC, Pretoria. I-xvii + 8661pp
Austen JM, Barbosa AD (2021) Diversity and epidemiology of bat trypanosomes: a one health perspective. Pathogens 10:1148. https://doi.org/10.3390/pathogens10091148
Beltz LA (2017) Kinetoplastids and bats. In: Bats and human health. Wiley Blackwell, Hoboken, pp 285–301
Büscher P, Cecchi G, Jamonneau V et al (2017) Human African trypanosomiasis. The Lancet 390:2397–2409. https://doi.org/10.1016/S0140-6736(17)31510-6
Clement L, Dietrich M, Markotter W et al (2020) Out of Africa: the origins of the protozoan blood parasites of the Trypanosoma cruzi clade found in bats from Africa. Mol Phylogenet Evol 145:106705. https://doi.org/10.1016/j.ympev.2019.106705
Darriba D, Posada D, Kozlov AM et al (2020) ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol 37:291–294. https://doi.org/10.1093/molbev/msz189
de Souza NN, Ursine RL, Cruz DS et al (2023) Leishmania species infection of bats: a systematic review. Acta Trop 248:107025. https://doi.org/10.1016/j.actatropica.2023.107025
Gardner RA, Molyneux DH (1987) Babesia vesperuginis: natural and experimental infections in British bats (Microchiroptera). Parasitology 95:461–469. https://doi.org/10.1017/s0031182000057887
Garnham PC, Heisch RB, Minter DM (1961) The vector of Hepatocystis (Plasmodium) kochi; the successful conclusion of observations in many parts of tropical Africa. Trans R Soc Trop Med Hyg 55:497–502. https://doi.org/10.1016/0035-9203(61)90071-2
Hamilton PB, Cruickshank C, Stevens JR et al (2012) Parasites reveal movement of bats between the New and Old Worlds. Mol Phylogenet Evol 63:521–526. https://doi.org/10.1016/j.ympev.2012.01.007
Kamani J, Atuman YJ, Oche DA et al (2022) Molecular detection of Trypanosoma spp. and Hepatocystis parasite infections of bats in Northern Nigeria. Parasitology 149:1460–1467. https://doi.org/10.1017/S0031182022000890
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. https://doi.org/10.1093/nar/gkf436
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Lima L, Silva FM, Neves L et al (2012) Evolutionary insights from bat trypanosomes: morphological, developmental and phylogenetic evidence of a new species, Trypanosoma (Schizotrypanum) erneyi sp. nov., in African bats closely related to Trypanosoma (Schizotrypanum) cruzi and allied species. Protist 163:856–872. https://doi.org/10.1016/j.protis.2011.12.003
Lima L, Espinosa-Alvarez O, Hamilton PB et al (2013) Trypanosoma livingstonei: a new species from African bats supports the bat seeding hypothesis for the Trypanosoma cruzi clade. Parasit Vectors 6:221. https://doi.org/10.1186/1756-3305-6-221
Morrison LJ, Vezza L, Rowan T et al (2016) Animal African trypanosomiasis: time to increase focus on clinically relevant parasite and host species. Trends Parasitol 32:599–607. https://doi.org/10.1016/j.pt.2016.04.012
Njiokou F, Laveissière C, Simo G et al (2006) Wild fauna as a probable animal reservoir for Trypanosoma brucei gambiense in Cameroon. Infect Genet Evol 6:147–153. https://doi.org/10.1016/j.meegid.2005.04.003
Noyes H, Stevens JR, Teixeira M et al (1999) A nested PCR for the ssrRNA gene detects Trypanosoma binneyi in the platypus and Trypanosoma sp. in wombats and kangaroos in Australia. Int J Parasitol 29:331–339. https://doi.org/10.1016/S0020-7519(98)00167-2
Patterson BD, Webala PW (2012) Keys to the bats (Mammalia: Chiroptera) of East Africa. Fieldiana Life Earth Sci 6:1–60
Rosevear DR (1965) The bats of West Africa. London: trustees of the British Museum (Natural History)
Schaer J, Perkins SL, Decher J et al (2013) High diversity of West African bat malaria parasites and a tight link with rodent Plasmodium taxa. Proc Natl Acad Sci 110:17415–17419. https://doi.org/10.1073/pnas.1311016110
Schaer J, Reeder DM, Vodzak ME et al (2015) Nycteria parasites of Afrotropical insectivorous bats. Int J Parasitol 45:375–384. https://doi.org/10.1016/j.ijpara.2015.01.008
Simpson AGB, Stevens JR, Lukeš J (2006) The evolution and diversity of kinetoplastid flagellates. Trends Parasitol 22:168–174. https://doi.org/10.1016/j.pt.2006.02.006
Szentivanyi T, Markotter W, Dietrich M et al (2020) Host conservation through their parasites: molecular surveillance of vector-borne microorganisms in bats using ectoparasitic bat flies. Parasite 27:72. https://doi.org/10.1051/parasite/2020069
Thiombiano NG, Boungou M, Chabi BAM et al (2023) First investigation of blood parasites of bats in Burkina Faso detects Hepatocystis parasites and infections with diverse Trypanosoma spp. Parasitol Res 122:3121–3129. https://doi.org/10.1007/s00436-023-08002-2
Tsague KJA, Bakwo Fils EM, Atagana JP et al (2022) Hepatocystis and Nycteria (Haemosporida) parasite infections of bats in the central region of Cameroon. Parasitology 149:51–58. https://doi.org/10.1017/S0031182021001542
Acknowledgements
We thank François Nguetsop for the coaching in microscopic work.
Funding
Open Access funding enabled and organized by Projekt DEAL. JS is funded by an individual research grant from the German Research Foundation (DFG; project number 437846632). The field work was supported by the special funds from the Cameroon government allocated for the modernization of research in the state universities (T Tchuinkam and EM Bakwo-Fils), through the Presidential Decret No: 2009/121 of 08 April 2009.
Author information
Authors and Affiliations
Contributions
KT and TT conceived and designed the study. KT, DM, JA, and EBF carried out field work and bat sampling and data gathering. LP and JS performed molecular work and JS performed phylogenetic analysis. KT and JS wrote the manuscript with input and editing from all authors.
Corresponding authors
Ethics declarations
Ethics approval
All surveys were reviewed and approved by the authorization from the Ministry of Scientific Research and Innovation under permit No 0000039/MINRESI/BOO/COO/C10/C11 from 16 February 2016. All work was performed in accordance with the relevant guidelines and regulations regarding care and use of animals.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Julia Walochnik
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
T. Tchuinkam and J. Schaer shared last authors.
Key findings
• Detection of Trypanosoma parasites in six bat species in Central Cameroon.
• Putative new subspecies in Trypanosoma livingstonei species group.
• Co-infections of trypanosomes and haemosporidian parasites in two bat species.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsague, K.J.A., Bakwo Fils, E.M., Atagana, J.P. et al. Molecular detection of trypanosomes of the Trypanosoma livingstonei species group in diverse bat species in Central Cameroon. Parasitol Res 123, 280 (2024). https://doi.org/10.1007/s00436-024-08303-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00436-024-08303-0