
Abstract
Bats are hosts to a large diversity of eukaryotic protozoan blood parasites that comprise species of Trypanosoma and different haemosporidian parasite taxa and bats have played an important role in the evolutionary history of both parasite groups. However, bats in several geographical areas have not been investigated, including in Burkina Faso, where no information about malaria parasites and trypanosomes of bats exists to date.
In this study, we collected data on the prevalence and the phylogenetic relationships of protozoan blood parasites in nine different bat species in Burkina Faso. Hepatocystis parasites were detected in two species of epauletted fruit bats, and a relatively high diversity of trypanosome parasites was identified in five bat species. The phylogenetic analyses recovered the trypanosome parasites of the bat species Rhinolophus alcyone and Nycteris hispida as close relatives of T. livingstonei, the trypanosome infections in Scotophilus leucogaster as closely related to the species T. vespertilionis and the trypanosomes from Pipistrellus nanulus and Epomophorus gambianus might present the species T. dionisii. These findings of the first investigation in Burkina Faso present a first snapshot of the diversity of protozoan blood parasites in bats in this country.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bats (order Chiroptera) represent up to 20% of all mammalian species with 1470 species described to date (Simmons and Cirranello 2023). They are considered key organisms in maintaining the ecosystem balance as they play essential roles in regulating insect populations, pollination, and seed dispersal (e.g., Kalka et al. 2008; Kunz et al. 2011; Aziz et al. 2021). Bats are hosts to a large diversity of eukaryotic protozoan blood parasites that comprise species of Trypanosoma and different haemosporidian parasite taxa and several studies suggest that bats have played an important role in the evolutionary history of both parasite groups (e.g., Duval et al. 2007; Hamilton et al. 2012; Lima et al. 2012, 2013; Schaer et al. 2013; Perkins and Schaer 2016; Galen et al. 2018; Clement et al. 2020; Austen and Barbosa 2021).
Trypanosomes (Kinetoplastea: Trypanosomatidae) are protozoan blood parasites that are distributed across all continents. They have adapted to infect all classes of vertebrates, feature complex life cycles and are primarily transmitted to vertebrate hosts through blood-feeding arthropods or leeches (e.g., Simpson et al. 2006). Different Trypanosoma species pose threats to both humans and livestock; in Africa these comprise the three Trypanosoma species Trypanosoma brucei sensu lato, Trypanosoma vivax and Trypanosoma congolense (Morrison et al. 2016; Büscher et al. 2017). Bats are known to harbor a wide range of Trypanosoma species; however, our understanding of species diversity, vectors, life cycles, distribution, and the evolutionary history of bat trypanosomes remains limited (e.g., Hamilton et al. 2012; Lima et al. 2012, 2013; Clement et al. 2020; Austen and Barbosa 2021).
Haemosporidian parasites, belonging to the phylum Apicomplexa, infect a wide array of birds, saurian reptiles, and mammals (Garnham 1966). The Plasmodium species that infect humans and cause the malaria disease are part of a large group of haemosporidian parasites, encompassing approximately 500 closely related species (Martinsen and Perkins 2013; Galen et al. 2018). These parasites utilize a diverse range of dipteran and vertebrate hosts to complete their life cycles (Garnham 1966; Levine 1988). Mammalian haemosporidian parasites are classified into different genera, including Plasmodium, Hepatocystis, Polychromophilus and Nycteria (Perkins and Schaer 2016). Among mammals, bats seem to feature an exceptionally high diversity of haemosporidian parasites and comprehensive sampling and systematic analysis of bat malaria parasites are vital for gaining a better understanding of the evolutionary history of haemosporidian parasites, including those that infect humans (e.g., Garnham 1966; Schaer et al. 2013; Perkins and Schaer 2016).
Despite the large haemosporidian and trypanosome parasite diversity being reported from bats, data from several geographical areas have not been collected yet. One of these “blind spots” is in Burkina Faso where no information about haemosporidian parasites and trypanosomes of bats exists to date. With about 52 species of bats, Burkina Faso features a diverse fauna of bats (Kangoyé et al. 2012, 2015; Thiombiano et al. 2019).
This study presents data on the prevalence and the phylogenetic relationships of protozoan blood parasites in different bat species in Burkina Faso.
Materials and methods
Field sampling and microscopy
Bats were sampled in Burkina Faso in December 2020 (wet season), January to April 2021 (dry season) and August 2021 (wet season) (Table S1). Burkina Faso is a Sahelian country located in West Africa. Its population was estimated at 21,509,443 in 2021 (INSD, 2021). Burkina Faso is characterized by a tropical climate alternating between a long dry season (October to April) and a short rainy season (May to September). Sampling was carried out at five different sampling sites that were characterized by the presence of fruit tree orchards (e.g., Mango trees), and only the sampling site in Diebougou was located close to a cave (Fig. 1).

Map of Burkina Faso. Sampling sites are depicted by dots. The sampling sites Bama, Banzon, Kiri and Ouagadougou are characterized by the presence of fruit tree orchards (e.g., Mango trees), while the sampling site in Diebougou is located close to a cave
Bats were captured using mist nets. The nets were set every capture night between 5 pm and 5 am. The bats were gently removed from the nets and transferred into individual bags. Measurements were taken to identify the bats to genus and/or species level using different identification keys (Rosevear 1965; Hayman 1971; Bergmans 1997). For the investigation of blood parasites, small blood samples were collected by venous puncture of the brachial vein and collected in capillary tubes. These blood samples were used to spot blood dots onto DNA FTA cards for subsequent molecular analysis and to prepare thin blood smears on slides for microscopic analysis. All bats were released at the capture sites, and it was assured that the bleeding had stopped. The thin blood smears were dried and fixed in 99–100% (vol/vol) ethanol solution for 3 s in the field and fixed again with 100% (vol/vol) methanol in the laboratory (in Germany), before staining with 10% Giemsa solution for about 40 min. Giemsa-stained blood smears were examined for the presence of haemosporidian and trypanosome parasites using light microscopy at a magnification of × 400 and × 1000 with immersion oil.
Molecular methods and phylogenetic analysis
Whole genomic DNA was extracted from the dried blood dots on DNA FTA cards using the DNeasy extraction kit (Qiagen, Hilden, Germany) (e.g., Schaer et al. 2017). The protocol for animal tissues was performed and samples were eluted in 80–100 µl AE buffer. PCRs were performed using the AllTa q Master Mix Kit (QIAGEN) with 4–5 µl of genomic DNA as the template, and 1 µl of each primer (10 mM). For the detection and phylogenetic analysis of haemosporidian parasites (Plasmodium, Hepatocystis, Nycteria, Polychromophilus), the mitochondrial genes cytochrome b (cytb) and cytochrome oxidase 1 (cox1) and the nuclear gene elongation factor 2 (ef2) of the parasites were amplified and Sanger-sequenced (following e.g., Schaer et al. 2013). To detect and characterize trypanosome infections, a nested-PCR approach was employed and about 640 bp of the small subunit 18S ribosomal RNA gene (18S rRNA) was amplified (using the approach described by Noyes et al. 1999). Following this, for all the samples that tested positive, a second gene, the nuclear glycosomal glyceraldehyde phosphate dehydrogenase (gGAPDH), was targeted using a nested-PCR approach (as outlined by Clement et al. 2020). All primers are listed in Table S2. All positive PCR products were sequenced with the amplification primers and run on an ABI-373 sequencer. The software programs Geneious Prime 2023.1.2 (https://www.geneious.com) and MEGA 11.0.10 (https://www.megasoftware.net) were used to quality-check and manually edit all nucleotide sequences. Instances of double nucleotide peaks (where both peaks have a minimum height of 40% at a single nucleotide position) observed in the sequence electropherograms of sequence segments with high quality within individual sequence assemblies were documented as indicative of mixed haplotype infections. Amplification and sequencing were repeated for samples with sequences of lower quality. Individual sequences per sample were assembled and exported as a consensus sequence for subsequent analysis. Sequences were aligned using the MAFFT algorithm (Katoh et al. 2002; Katoh and Standley 2013). GenBank was utilized to obtain reference sequences, which were subsequently added to the alignments. Accession numbers for the analysis of haemosporidian parasites are provided in Table S3, while the accession numbers for the 18S rRNA and gGAPDH trypanosome analyses are provided in the respective phylogenetic tree figures and Table S4. The concatenated alignment of three genes for the analysis of the phylogenetic relationships of haemosporidian parasites comprised 123 sequences (including sequences from 16 representative samples of this study) and had a total length of 1938 nucleotides (nt) (414 nt of ef2 gene, 531 nt of cytb, 993 nt of cox1). The alignment of cytb for phylogenetic analysis of Hepatocystis parasites in African bats comprised 82 sequences and had a length of 531 nt. Sequence alignments for the analysis of Trypanosoma spp. included 89 sequences and a length of 726 nt for 18S rRNA and 79 sequences and 894 nt for gGAPDH respectively. Data was partitioned according to genes (for the protein-coding genes) and the software modeltest-ng 0.1.7 (Darriba et al. 2020), implemented in raxmlGUI version 2.0.10 (Edler et al. 2021) was used to test different DNA substitution models. Maximum Likelihood (ML) analyses were conducted using raxmlGUI 2.0.10 to evaluate phylogenetic relationships. The haemosporidian parasite analysis of the concatenated dataset was carried out using the substitution model GTR + I (proportion of invariant) + Gamma (rate heterogeneity) and the taxon Leucocytozoon as outgroup and the nodal support was evaluated using 1000 replicates (thorough bootstrap). The Hepatocystis parasite analysis of the cytb dataset was carried out using the substitution model TIM2 + I (proportion of invariant) and the taxon Plasmodium falciparum as outgroup and the nodal support was evaluated using 1000 replicates (thorough bootstrap). The Maximum Likelihood analyses of the trypanosome datasets of the 18S rRNA and gGAPDH gene were carried out using the models TIM3 + I + G and GTR + I + G (with 10,000 thorough bootstrapping) respectively and the outgroup taxon Trypanosoma lewisi (following Kamani et al. 2022). All resulting phylogenetic trees were displayed in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
Prevalence of protozoan blood parasites
A total of 119 bats belonging to six bat families, seven genera and nine species were investigated. Screening with microscopy and PCR methods detected haemosporidian and trypanosome parasite infections in bats of Burkina Faso (Table 1; Table S1). Haemosporidian parasites were recorded in 26 bat individuals (26/119), corresponding to a prevalence of 21.8%. These parasites, identified as species of the genus Hepatocystis, were detected in individuals of the two bat species Epomophorus gambianus and Epomophorus pusillus (bat family Pteropodidae) that were sampled in the two locations of Bama and Ouagadougou. All ten E. pusillus individuals were captured in the wet season and featured infections with Hepatocystis parasites (10/10, 100%). The prevalence of infections in E. gambianus was 41,1% (16/38), with 10% (4/38) in samples from the dry season and a higher prevalence of 32% (12/38) in the individuals that got captured in the wet season. Although previous research has identified Nycteria and Polychromophilus parasites in bat species belonging to the Nycteridae, Rhinolophidae, and Vespertilionidae, we did not detect haemosporidian infections in bats of these bat families in this study (Table 1).
Trypanosoma spp. parasites were detected in 12 individuals (12/119) corresponding to an overall prevalence of 10.1% in individuals of five different bat species belonging to four bat families captured in the three locations Bazon, Diebougou and Ouagadougou (Table 1). Trypanosome infections were recorded in one individual per species in Epomophorus gambianus (1/38 = 2.6%), Nycteris hispida (1/1 = 100%) and Pipistrellus nanulus (1/10 = 10%). Prevalences of 36% and 17% were recorded for Rhinolophus alcyone (4/11 = 36.4%) and Scotophilus leucogaster (5/29 = 17.2%) respectively.
Characterization of Hepatocystis parasites of bats in Burkina Faso
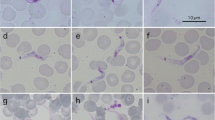
The quality of the blood smears did not permit a detailed evaluation of the morphology of the gametocyte blood stages of the Hepatocystis parasites of E. pusillus and E. gambianus. However, as characteristic for Hepatocystis parasites, the detected blood stages were limited to gametocyte stages (mostly mature stages) and hemozoin pigment was present in all parasite cells in the infected erythrocytes (e.g., Garnham 1966; Schaer et al. 2013; Atama et al. 2019) (Fig. S1). High quality sequences for at least one molecular marker could be generated for 25 of the 26 samples. For one sample (isolate Tengo1) only a low-quality sequence for the partial cox1 gene was available that however shared highest identity with Hepatocystis sp. of African bats in NCBI BLASTn search (e.g., with NCBI accession number MZ460952) and was thus scored as infected with Hepatocystis sp. All Hepatocystis infections in E. pusillus and E. gambianus of this study were determined as mixed haplotype infections as double nucleotide peaks were recorded in the sequences of all samples. As haplotype network analysis can only be performed with single haplotype infections, the number of haplotypes present in our sample set could not be determined. However, the phylogenetic analysis confirmed the parasites of E. pusillus und E. gambianus from Burkina Faso as Hepatocystis sp. which group in the main Hepatocystis clade that contains all other African bat Hepatocystis parasites (Fig. 2). Within this clade, the Hepatocystis parasite sequences of this study (highlighted in blue) fall in different places and do not cluster in host genus or species-specific clades just like the bat Hepatocystis sequences from other countries and locations in both West Africa and Central-/East Africa that exhibit no pattern of clustering according to countries or host species (Fig. 2, Fig. S2).

Maximum likelihood analysis of Hepatocystis parasites in African bat species. A The analysis is based on the concatenated dataset of the parasites mitochondrial genes cytb (531nt) and cox1 (993nt) and one nuclear gene (414 nt of ef2 gene) and was run in the context of the major haemosporidian parasite clades Haemoproteus, Parahaemoproteus, Polychromophilus and the mammalian-infecting Plasmodium clades. Leucocytozoon was used as outgroup taxon. The phylogenetic analysis recovered the Hepatocystis sequences from E. pusillus and E. gambianus of this study within the African bat Hepatocystis clade (collapsed in A). B Section of the African bat Hepatocystis clade (uncollapsed). No strict clustering of African bat Hepatocystis sequences according to country or host species is apparent which is in line with previous findings (e.g., Schaer et al. 2017). Sequences of the study from Burkina Faso are highlighted in blue. Numbers at nodes are ML bootstrap values (> 50) using 1000 replicates. The photograph depicts the bat host species E. gambianus
Molecular characterization of Trypanosoma spp. parasites of bats in Burkina Faso
Three of the four trypanosome samples of R. alcyone (Rhinolophidae) share identical 18S rRNA sequences and thus represent one haplotype (isolates DIE15, DIE16, DIE22). The 18S rRNA phylogenetic analysis recovered this haplotype within the T. livingstonei clade. Close relatives are T. livingstonei parasites that were isolated from different bat species in Africa (Hipposideros caffer, Mozambique, KF192984; Rhinolophus landeri, Mozambique, KF192982 and Rhinolophus simulator, South Africa, MN956683) (Fig. 3). The sequence of the fourth trypanosome sample from R. alcyone of this study (isolate DIE19) features highest sequence similarity with a Trypanosoma sp. reported from Eidolon helvum in Nigeria. Together these two sequences group with a Trypanosoma sp. clade that comprises Trypanosoma parasite sequences from different Miniopterus bat hosts and bat flies from Europe and South Africa (Szentivanyi et al. 2020) and this whole clade shares a close relationship with T. livingstonei parasites (Fig. 3). The trypanosome parasite detected in N. hispida shares highest sequence identity with a reference sequence of a trypanosome isolated from Nycteris macrotis from Nigeria (ON326586) which together also group within the T. livingstonei/Trypanosoma cf. livingstonei clade. Unfortunately, no gGAPDH sequences could be successfully amplified for the trypanosomes of R. alcyone and N. hispida.

Maximum likelihood phylogeny of the Trypanosoma parasites based on the 18S rRNA alignment. The alignment featured a total length of 726 bp and the evolutionary model TIM3 + I + G was used for the phylogenetic analysis and the taxon Trypanosoma lewisi as outgroup. Numbers at nodes are ML bootstrap values using 10,000 replicates. The representative sequence for the haplotype 1 recovered in three R. alcyone samples groups within the T. livingstonei clade, the sequence of the fourth trypanosome sample from R. alcyone of this study (isolate DIE19) features highest sequence similarity with a Trypanosoma sp. from Eidolon helvum in Nigeria. The trypanosome parasite detected in N. hispida shares highest sequence identity with a trypanosome isolated from Nycteris macrotis from Nigeria which together group within the T. livingstonei/Trypanosoma cf. livingstonei clade. All trypanosome sequences of R. alcyone and N. hispida are highlighted in bold green. The trypanosome sequences from the parasites isolated from S. leucogaster (highlighted in bold red) group in a clade that comprises Trypanosoma vespertilionis sequences. The trypanosome sequences from P. nanulus and E. gambianus group with T. dionisii sequences recovered from diverse bat species (the gGAPDH sequences for both samples group within the T. dionisii clade with high support, see Fig. S3). The photograph depicts the bat host species R. alcyone
The trypanosome parasites in the five bat samples of S. leucogaster share highest identities with sequences of the species Trypanosoma vespertilionis. The 18S rRNA sequences of four samples (isolates BAZ5, BAZ6, BAZ11, BAZ21) represent one haplotype. The 18S rRNA phylogenetic analysis recovered this haplotype within T. vespertilionis sequences and with closest relationship with a T. vespertilionis reference of Scotophilus sp. from Guinea-Bissau (MF144889) (Fig. 3). The 18S rRNA of the fifth sample was of low quality. However, a high quality gGAPDH sequence for this sample was generated and the phylogenetic analysis confirmed this trypanosome sample of S. leucogaster (isolate BSOM18) as also belonging to the T. vespertilionis/T. cf. vespertilionis group (Fig. S3). The trypanosomes from P. nanulus and E. gambianus group with T. dionisii sequences from different bat species in the 18S rRNA analysis (Fig. 3) and further the gGAPDH sequences for both samples were recovered within the T. dionisii clade with high support (Fig. S3).
Unfortunately, no trypanosome parasite stages were detected in any of the blood smears of the bat hosts despite thorough screening, which points to very low parasitemia levels in all samples.
Discussion
This study provides the first information on haemosporidian and trypanosome parasites of bats in Burkina Faso. Hepatocystis parasites were detected in two bat species, while trypanosomes were verified from five bat species. Interestingly, no co-infections of haemosporidian parasites and trypanosomes were detected in the study samples.
Hepatocystis parasites were detected in two species of epauletted fruit bats confirming previous findings of Epomophorus spp. being common Hepatocystis sp. hosts across their African distribution range (Schaer et al. 2013; Lutz et al. 2016; Boundenga et al. 2018; Atama et al. 2019). The morphospecies Hepatocystis epomophori has been described from Epomophorus species in Africa (Rodhain 1926), but it has been suggested that the Hepatocystis parasites of African epauletted fruit bats represent several close related taxa or cryptic species (Schaer et al. 2017). Unfortunately, the quality of the blood smears did not allow an assignment of the Hepatocystis parasites of the study to any morphospecies. However, the phylogenetic analyses recovered the parasites among sequences of parasites previously determined as belonging to a Hepatocystis epomophori species complex which lack signatures of host specificity and do not strictly group to country origin within Africa (Schaer et al. 2017). The recorded prevalences were comparable to findings of other studies in epauletted fruit bats in Africa (e.g., Schaer et al. 2013; Lutz et al. 2016; Boundenga et al. 2018) and we observed a seasonal trend with higher prevalence in the wet compared to the dry season samples confirming previous findings (Schaer et al. 2017). Interestingly, all Hepatocystis infections in this study represented mixed haplotype infections which might point to repeated infections over time with different parasite haplotypes that circulate in the area. Mixed haplotype infections have been reported before in Hepatocystis infections in primates and African epauletted fruit bats (Thurber et al. 2013; Schaer et al. 2017). Subsequent investigations are important to study the dynamics of Hepatocystis parasite infections in these bat hosts in Burkina Faso in more detail.
A relatively high diversity of trypanosome parasites was identified in the bat species of the study. Parasites that might represent T. livingstonei were recorded in the insectivorous bat species R. alcyone and N. hispida. Infections with T. livingstonei and T. cf. livingstonei seem to be prevalent in various African insectivorous bat species (Lima et al. 2013; Clement et al. 2020). The trypanosome infections in S. leucogaster feature closest phylogenetic relationships with the species T. vespertilionis, a species that has been reported from diverse European and African bats (Stevens et al. 1998; Espinosa-Álvarez et al. 2018; Clement et al. 2020). The trypanosomes from P. nanulus and E. gambianus share closest relationships with the species T. dionisii, a trypanosome species that is globally distributed and has been described in bats from the Americas, Europe, Africa and more recently also in Asian and Australian bats (e.g., Hamilton et al. 2012; Lima et al. 2013; Hodo et al. 2016; Espinosa-Álvarez et al. 2018; Mafie et al. 2018; Wang et al. 2019; Austen et al. 2020). These findings confirm the notion that African bat trypanosomes exhibit high phylogenetic diversity, potentially harboring a range of yet undiscovered species (Clement et al. 2020). Investigating the diversity and the phylogenetic relationships of bat trypanosomes is crucial for enhancing our understanding of the whole group of trypanosomes, as some species are a threat to humans (Hamilton et al. 2009; Lima et al. 2012). Consequently, a more targeted systematic sampling and subsequent molecular characterization of trypanosome species from African bats is needed.
The current study only represents a snapshot of the diversity of protozoan bat blood parasites in Burkina Faso as only nine of the 52 bat species of Burkina Faso have been investigated. Further investigations targeting a broader taxon sampling with larger sample sizes collected across different seasons will provide a more complete picture.
Data availability
All Hepatocystis and trypanosome sequences of the study are available at GenBank (NCBI) with the accession numbers OR462727-OR462732, OR469745-OR469791.
References
Atama N, Manu S, Ivande S et al (2019) Survey of Hepatocystis parasites of fruit bats in the Amurum forest reserve, Nigeria, identifies first host record for Rousettus aegyptiacus. Parasitology 146:1550–1554. https://doi.org/10.1017/S0031182019000817
Austen JM, Barbosa AD (2021) Diversity and epidemiology of bat trypanosomes: a one health perspective. Pathogens 10:1148. https://doi.org/10.3390/pathogens10091148
Austen JM, Van Kampen E, Egan SL et al (2020) First report of Trypanosoma dionisii (Trypanosomatidae) identified in Australia. Parasitology 147:1801–1809. https://doi.org/10.1017/S0031182020001845
Aziz SA, McConkey KR, Tanalgo K et al (2021) The critical importance of old-world fruit bats for healthy ecosystems and economies. Frontiers in Ecology and Evolution 9:641411. https://doi.org/10.3389/fevo.2021.641411
Bergmans W (1997) Taxonomy and biogeography of African fruit bats (Mammalia, Megachiroptera). 5. The genera Lissonycteris Andersen, 1912, Myonycteris Matschie, 1899 and Megaloglossus Pagenstecher, 1885; general remarks and conclusions; annex: key to all species. Beaufortia 47:11–90
Boundenga L, Ngoubangoye B, Mombo IM et al (2018) Extensive diversity of malaria parasites circulating in Central African bats and monkeys. Ecol Evol 8:10578–10586. https://doi.org/10.1002/ece3.4539
Büscher P, Cecchi G, Jamonneau V et al (2017) Human African trypanosomiasis. The Lancet 390:2397–2409. https://doi.org/10.1016/S0140-6736(17)31510-6
Clement L, Dietrich M, Markotter W et al (2020) Out of Africa: the origins of the protozoan blood parasites of the Trypanosoma cruzi clade found in bats from Africa. Mol Phylogenet Evol 145:106705. https://doi.org/10.1016/j.ympev.2019.106705
Darriba D, Posada D, Kozlov AM et al (2020) ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol 37:291–294. https://doi.org/10.1093/molbev/msz189
Duval L, Robert V, Csorba G et al (2007) Multiple host-switching of Haemosporidia parasites in bats. Malar J 6:157. https://doi.org/10.1186/1475-2875-6-157
Edler D, Klein J, Antonelli A, Silvestro D (2021) raxmlGUI 2.0: a graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol Evol 12:373–377. https://doi.org/10.1111/2041-210X.13512
Espinosa-Álvarez O, Ortiz PA, Lima L et al (2018) Trypanosoma rangeli is phylogenetically closer to Old World trypanosomes than to Trypanosoma cruzi. Int J Parasitol 48:569–584. https://doi.org/10.1016/j.ijpara.2017.12.008
Galen SC, Borner J, Martinsen ES et al (2018) The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order Haemosporida) reveal widespread taxonomic conflict. Royal Society Open Science 5:171780. https://doi.org/10.1098/rsos.171780
Garnham PC (1966) Malaria parasites and other Haemosporida. Blackwell Scientific Publications, Oxford, pp 635–661
Hamilton PB, Adams ER, Njiokou F et al (2009) Phylogenetic analysis reveals the presence of the Trypanosoma cruzi clade in African terrestrial mammals. Infect Genet Evol 9:81–86. https://doi.org/10.1016/j.meegid.2008.10.011
Hamilton PB, Cruickshank C, Stevens JR et al (2012) Parasites reveal movement of bats between the New and Old Worlds. Mol Phylogenet Evol 63:521–526. https://doi.org/10.1016/j.ympev.2012.01.007
Hayman RWHJ (1971) Order Chiroptera. In: Meester J, Setzer HW (eds) The mammals of Africa, an identification manual. Smithsonian Institution, Washington, D.C., pp 1–73p
Hodo CL, Goodwin CC, Mayes BC et al (2016) Trypanosome species, including Trypanosoma cruzi, in sylvatic and peridomestic bats of Texas, USA. Acta Trop 164:259–266
Kalka MB, Smith AR, Kalko EKV (2008) Bats limit arthropods and herbivory in a tropical forest. Science 320:71–71. https://doi.org/10.1126/science.1153352
Kamani J, Atuman YJ, Oche DA et al (2022) Molecular detection of Trypanosoma spp. and Hepatocystis parasite infections of bats in Northern Nigeria. Parasitology 149:1460–1467. https://doi.org/10.1017/S0031182022000890
Kangoyé NM, Oueda A, Oueda A et al (2012) Bats (Chiroptera) of Burkina Faso: preliminary list with fifteen first record species. Int J Bio Chem Sci 6:6017–6030. https://doi.org/10.4314/ijbcs.v6i6.29
Kangoyé NM, Ouéda A, Granjon L et al (2015) Diversity and distribution of bats (Mammalia Chiroptera) in Burkina Faso. Biodiversity Journal 6:597–632
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. https://doi.org/10.1093/nar/gkf436
Kunz TH, Braun de Torrez E, Bauer D et al (2011) Ecosystem services provided by bats. Ann N Y Acad Sci 1223:1–38. https://doi.org/10.1111/j.1749-6632.2011.06004.x
Levine N (1988) The protozoan phylum Apicomplexa. CRC Press
Lima L, Silva FM, Neves L et al (2012) Evolutionary insights from bat trypanosomes: morphological, developmental, and phylogenetic evidence of a new species, Trypanosoma (Schizotrypanum) erneyi sp. nov., in African bats closely related to Trypanosoma (Schizotrypanum) cruzi and allied species. Protist 163:856–872. https://doi.org/10.1016/j.protis.2011.12.003
Lima L, Espinosa-Alvarez O, Hamilton PB et al (2013) Trypanosoma livingstonei: a new species from African bats supports the bat seeding hypothesis for the Trypanosoma cruzi clade. Parasit Vectors 6:221. https://doi.org/10.1186/1756-3305-6-221
Lutz HL, Patterson BD, Kerbis Peterhans JC et al (2016) Diverse sampling of East African haemosporidians reveals chiropteran origin of malaria parasites in primates and rodents. Mol Phylogenet Evol 99:7–15. https://doi.org/10.1016/j.ympev.2016.03.004
Mafie E, Rupa FH, Takano A et al (2018) First record of Trypanosoma dionisii of the T. cruzi clade from the Eastern bent-winged bat (Miniopterus fuliginosus) in the far East. Parasitol Res 117:673–680
Martinsen ES, Perkins SL (2013) The diversity of Plasmodium and other haemosporidians: the intersection of taxonomy, phylogenetics, and genomics. In: Carlton JM, Perkins SL, Deitsch KW (eds) Malaria parasites: comparative genomics, evolution, and molecular biology. Caister Academic Press, Poole, Uk, pp 1–15
Morrison LJ, Vezza L, Rowan T et al (2016) Animal African trypanosomiasis: time to increase focus on clinically relevant parasite and host species. Trends in Parasitology 32:599–607. https://doi.org/10.1016/j.pt.2016.04.012
Noyes H, Stevens JR, Teixeira M et al (1999) A nested PCR for the ssrRNA gene detects Trypanosoma binneyi in the platypus and Trypanosoma sp. in wombats and kangaroos in Australia. Int J Parasitol 29:331–339. https://doi.org/10.1016/S0020-7519(98)00167-2
Perkins SL, Schaer J (2016) A modern menagerie of mammalian malaria. Trends Parasitol 32:772–782. https://doi.org/10.1016/j.pt.2016.06.001
Rodhain J (1926) Plasmodium epomophori n. sp. parasite commun des Roussettes epaulieres au Congo Belge. Bull Soc Pathol Exot Filiales 19:828–838
Rosevear DR (1965) The bats of West Africa. London: trustees of the British Museum (Natural History).
Schaer J, Perkins SL, Decher J et al (2013) High diversity of West African bat malaria parasites and a tight link with rodent Plasmodium taxa. Proc Natl Acad Sci 110:17415–17419. https://doi.org/10.1073/pnas.1311016110
Schaer J, Perkins SL, Ejotre I et al (2017) Epauletted fruit bats display exceptionally high infections with a Hepatocystis species complex in South Sudan. Sci Rep 7:6928. https://doi.org/10.1038/s41598-017-07093-z
Simpson AGB, Stevens JR, Lukeš J (2006) The evolution and diversity of kinetoplastid flagellates. Trends in Parasitology 22:168–174. https://doi.org/10.1016/j.pt.2006.02.006
Simmons NB and Cirranello AL (2023) Bat species of the world: a taxonomic and geographic database. Version 1.4. https://batnames.org/home.html. Accessed 20 Jul 2023
Stevens J, Noyes HA, Dover GA et al (1998) The ancient and divergent origins of the human pathogenic trypanosomes, Trypanosoma brucei and Trypanosoma cruzi. Parasitology 18:107–116
Szentivanyi T, Markotter W, Dietrich M et al (2020) Host conservation through their parasites: molecular surveillance of vector-borne microorganisms in bats using ectoparasitic bat flies. Parasite 27:72. https://doi.org/10.1051/parasite/2020069
Thiombiano NG, Boungou M, Kangoyé NM et al (2019) Inventory of bat (Scotophilus leucogaster, Cretzschmar 1826) ectoparasites of savannah area in Burkina Faso. J Bio Env Sci 15:109–116
Thurber MI, Ghai RR, Hyeroba D et al (2013) Co-infection and cross-species transmission of divergent Hepatocystis lineages in a wild African primate community. Int J Parasitol 43:613–619. https://doi.org/10.1016/j.ijpara.2013.03.002
Wang LJ, Han HJ, Zhao M et al (2019) Trypanosoma dionisii in insectivorous bats from northern China. Acta Trop 193:124–128
Acknowledgements
We thank Dr. NM Kangoyé for the help in bat species identification. We further thank Madou Sanou, Joel Guigma and Abdoul Razar Simpore for their assistance during field sampling.
Funding
Open Access funding enabled and organized by Projekt DEAL. NGT was supported by a Flexible Travel Fund by the Berlin Center for Global Engagement at the Berlin University Alliance (BUA) and is funded by an individual research grant from the International Funds for Science (IFS; project number I1-B-6714–1). JS is funded by an individual research grant from the German Research Foundation (DFG; project number 437846632).
Author information
Authors and Affiliations
Contributions
NGT and JS conceived and designed the study. NGT, MB, BAMC and AO carried out field work and bat sampling. NGT, JS, OW performed molecular work and phylogenetic analysis. All authors conducted data gathering and wrote the article.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Ministry of the Environment, Green Economy, and Climate Change (MEECVCC) (permit no. 21 -091 MEECVCC/SG/DGEF/DFRC). It was approved by the ethics committee on animal experimentation of Joseph Ki-Zerbo University with the number: no CEEA-UJKZ/2021–04. All work was performed in accordance with the relevant guidelines and regulations regarding care and use of animals.
Competing interests
The authors declare no competing interests.
Additional information
Section Editor: Alexander Maier.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key Findings
• First proof of Hepatocystis parasites in Epomophorus spp. in Burkina Faso.
• Detection of three Trypanosoma species in different bat species in Burkina Faso.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Thiombiano, N.G., Boungou, M., Chabi, B.A.M. et al. First investigation of blood parasites of bats in Burkina Faso detects Hepatocystis parasites and infections with diverse Trypanosoma spp.. Parasitol Res 122, 3121–3129 (2023). https://doi.org/10.1007/s00436-023-08002-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-023-08002-2