
Abstract
Plasmodium knowlesi is a simian malaria parasite that causes significant zoonotic infections in Southeast Asia, particularly in Malaysia. The Plasmodium thrombospondin-related apical merozoite protein (TRAMP) plays an essential role in the invasion of the parasite into its host erythrocyte. The present study investigated the genetic polymorphism and natural selection of the full length PkTRAMP from P. knowlesi clinical isolates from Malaysia. Blood samples (n = 40) were collected from P. knowlesi malaria patients from Peninsular Malaysia and Malaysian Borneo. The PkTRAMP gene was amplified using PCR, followed by cloning into a plasmid vector and sequenced. Results showed that the nucleotide diversity of PkTRAMP was low (π: 0.009). Z-test results indicated negative (purifying) selection of PkTRAMP. The alignment of the deduced amino acid sequences of PkTRAMP of Peninsular Malaysia and Malaysian Borneo revealed 38 dimorphic sites. A total of 27 haplotypes were identified from the amino acid sequence alignment. Haplotype analysis revealed that there was no clustering of PkTRAMP from Peninsular Malaysia and Malaysian Borneo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The World Health Organization (WHO) reported approximately 224 million malaria cases and 627,000 malaria death cases globally in 2020 (WHO 2021). Plasmodium knowlesi is endemic in Southeast Asia with the majority cases being reported from Malaysia (Luchavez et al. 2008; Ng et al. 2008; Van den Eede et al. 2009; Chin et al. 2020). Malaysia is vulnerable to transmission of malaria due to the tropical climate and its location in the humid equatorial region. In Malaysia, most of the cases of human P. knowlesi malaria have been reported in Malaysian Borneo. In the period 2013–2017, 77.1% of malaria cases in Malaysian Borneo were P. knowlesi malaria. Furthermore, the average malaria death rate in Malaysian Borneo is higher than in Peninsular Malaysia (Hussin et al. 2020). The reasons for the disparity in case numbers and severity between Peninsular Malaysia and Malaysian Borneo are unknown. However, genetically distinct haplotypes of some P. knowlesi genetic markers were detected in these two regions (Peninsular Malaysia and Malaysian Borneo) such as Duffy binding protein (PkDBPαII), cytochrome c oxidase subunit I (PkCOXI), type A small subunit ribosomal 18S RNA (PkA-type 18S rRNA), and apical membrane antigen-1 (PkAMA-1) (Fong et al. 2015; Yusof et al. 2016; Ng et al. 2021).
P. knowlesi malaria is a potentially life-threatening disease due to the parasite’s much shorter (i.e., 24 h) asexual erythrocytic stage development than other medically important malaria parasites (Knowles and Gupta 1932; Cox-Singh et al. 2008). The erythrocytic cycle of the parasite is responsible for the manifestation of symptoms suffered by malaria patients. The invasion of erythrocytes by Plasmodium merozoites involves multiple steps which includes initial attachment, followed by apical reorientation of the merozoite. A tight junction is subsequently formed, and the junction moves to the posterior of erythrocytes powered by actin-myosin motor. This is followed by the entry of the merozoite into a parasitophorous vacuole in the erythrocyte (Cowman and Crabb 2006).
The P. knowlesi thrombospondin-related apical merozoite protein (PkTRAMP) is a 360-amino acid invasion-related protein that is part of a protein family containing the thrombospondin structural homology repeat (TSR) domain (Thompson et al. 2004). These TSR-containing proteins are known to play a crucial role in cell adhesion and cell interactions during cell migration (Adams and Tucker 2000). The essential role of TRAMP in merozoite invasion and the parasite blood-stage development has been shown in previous studies, where TRAMP specific-induced antibodies have been shown to inhibit parasite invasion in vitro (Uchime et al. 2012; Siddiqui et al. 2013). However, the genetic diversity of TRAMP is yet to be fully investigated.
P. knowlesi is presently the main cause of human malaria infection in Malaysia, and there are higher case numbers and malaria mortality rate reported in Malaysian Borneo compared with Peninsular Malaysia. The aim of the present study, therefore, is to conduct a comparative analysis on the genetic polymorphism and natural selection of PkTRAMP in Malaysian P. knowlesi isolates. This is the first study on TRAMP sequences from P. knowlesi malaria clinical samples, and the findings will be beneficial in understanding the level of polymorphism in PkTRAMP for future functional and vaccine development studies.
Materials and methods
Human blood samples and DNA extraction
Blood samples from P. knowlesi malaria patients were collected from hospitals in the Peninsular Malaysia (n = 20) states of Johor, Pahang, Kedah, Johor, Kelantan, Negeri Sembilan, Perak, Terengganu, Selangor, and Federal Territory of Kuala Lumpur, as well as the Malaysian Borneo (n = 20) states of Sabah and Sarawak. Most of the blood samples for this study were obtained in 2019–2020. The presence of P. knowlesi in the samples was screened by microscopic examination of thick and thin blood smears and nested polymerase chain reaction (PCR) based on the Plasmodium 18S rRNA locus (Snounou et al. 1993; Imwong et al. 2009). Plasmodium DNA was extracted from the blood samples using the QIAGEN Blood and Tissue Kit (QIAGEN, Hilden, Germany). One hundred μl of blood was used for DNA extraction, and 50 μl of EB Buffer was used for DNA elution. The DNA was stored at -20 °C until use.
Polymerase chain reaction (PCR) amplification of PkTRAMP gene
PCR of the PkTRAMP gene was carried out using forward primer PkTRAMPfull-F: 5’-GGATCCATGCGGAGCTTCACCTTCATA-3’ and reverse primer PkTRAMPfull-R: 5’-GGATCCTTAATCGTACATAAATCATCCAGCCAC-3’. Approximately 0.5 μg of genomic DNA was used in a final amplification volume of 25 μl containing 2 mM MgCl2, 0.2 mM of dNTPs, 0.25 μM of forward and reverse primers, 1 unit of GoTaq® DNA polymerase, and 1X GoTaq® buffer (Promega, Madison, Wisconsin, USA). Cycling conditions for PCR were 95 °C for 3 min, followed by 35 cycles of 94 °C for 1 min, 53 °C for 1 min, and 72 °C for 100 s and a final extension at 72 °C for 10 min. The PCR products were subjected to electrophoresis on a 1% agarose gel stained with SYBR® Safe DNA gel stain (Invitrogen, Eugene, USA). PCR products were ligated into the pGEM-T® TA cloning vector (Promega, Madison, Wisconsin, USA), followed by transformation into Escherichia coli TOP10F’ competent cells using heat-shock method. The transformation colonies were screened for the presence of PkTRAMP DNA fragment. Positive recombinant plasmids were extracted and then sent to a commercial laboratory (Apical Scientific Sdn. Bhd., Malaysia) for DNA sequencing. For each isolate, three recombinant plasmid clones were sequenced to ensure that the PkTRAMP sequence obtained was consistent.
Genetic diversity analysis of PkTRAMP gene
DnaSP ver 6 (Rozas et al. 2017) was used to perform polymorphism analysis on the ~ 1020 bp sequences (n = 40). Information such as nucleotide diversity (π) and haplotype diversity (Hd) was calculated. The natural selection of PkTRAMP was determined by the Z-test (P < 0.05) in MEGA7 software based on Nei and Gojobori’s method with Jukes and Cantor correction (Nei and Gojobori 1986). Multiple nucleotide and deduced amino acid sequence alignments of generated PkTRAMP sequences including a reference sequence (P. knowlesi strain H, GenBank Accession Number XM_002262219) were performed using BioEdit sequence alignment editor ver 7.2.0. The haplotype network of PkTRAMP amino acid sequences was constructed using NETWORK program ver 4.6.1 (Bandelt et al. 1999).
Results
All newly generated PkTRAMP sequences (n = 40) from Peninsular Malaysia and Malaysian Borneo were deposited in the GenBank database under Accession Numbers ON892606-ON892645. The genetic diversity and natural selection pressure index of the full length PkTRAMP for whole Malaysia, Peninsular Malaysia, and Malaysian Borneo are presented in Table 1.
The overall nucleotide diversity of PkTRAMP from Malaysia was low (π = 0.009). The nucleotide diversity of PkTRAMP from Peninsular Malaysia and Malaysian Borneo was very similar (π = 0.008 and π = 0.007, respectively). Meanwhile, the overall haplotype diversity of PkTRAMP from Malaysia was high (Hd = 0.999). The haplotype diversity of PkTRAMP from Peninsular Malaysia and Malaysian Borneo was high (Hd = 1.000 and Hd = 0.995, respectively).
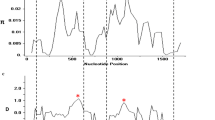
A sliding window plot with a window length of 100 bp and a step size of 25 bp was used to determine nucleotide diversity along the entire PkTRAMP sequence. The nucleotide diversity of PkTRAMP from Peninsular Malaysia and Malaysian Borneo ranged from 0.000 to 0.027 (Fig. 1a) and from 0.001 to 0.016 (Fig. 1b), respectively. For PkTRAMP from Peninsular Malaysia, the most conserved regions (π = 0.000) were within nucleotide positions 551–650, whereas the highest peak diversity (π = 0.027) was within nucleotide positions 826–925. In contrast, the same nucleotide position (826–925) was the most conserved region (π = 0.001) for PkTRAMP from Malaysian Borneo, whereas the highest peak diversity (π = 0.016) was within nucleotide positions 201–300. The Z-test revealed purifying (negative) selection on the PkTRAMP across Malaysia, Peninsular Malaysia, and Malaysian Borneo (dN < dS, P = 0.000).

Sliding window plot of the nucleotide diversity (π) along the PkTRAMP, generated with a window length of 100 bp and step size of 25 bp. a Nucleotide polymorphism in the PkTRAMP from Peninsular Malaysia. b Nucleotide polymorphism in the PkTRAMP from Malaysian Borneo
The PkTRAMP sequences (including strain H) were translated into amino acid sequences for analysis of haplotype polymorphism (Fig. 2). In the analysis, the PkTRAMP amino acid sequence of strain H was used as reference. Close examination identified 38 polymorphic sites, and all 38 were dimorphic. Overall, the amino acid sequences could be categorised into 27 haplotypes (H1-H27). Haplotype H1 had the highest frequency (n = 14), thus being the most predominant in the population. Haplotype network analysis (Fig. 3) of the PkTRAMP did not reveal any specific separation between Peninsular Malaysia and Malaysian Borneo. The largest node was H1, which contained members from both Peninsular Malaysia and Malaysian Borneo. All other haplotypes basically originated from H1.

Amino acid sequence polymorphism in PkTRAMP from Peninsular Malaysia and Malaysian Borneo. Polymorphic amino acid residues are listed for each haplotype. Amino acid residues identical to those of the reference sequence [strain H (haplotype 1)] are marked by dots. Dimorphic amino acid positions are marked in yellow shading. Haplotype frequency for each haplotype is listed in the right panel

Network analysis of 27 haplotypes of PkTRAMP. Red nodes indicate Malaysian Borneo haplotype members, and yellow nodes indicate Peninsular Malaysia haplotypes. The size of each node reflects the number of isolates in each haplotype
Discussion
In the current study, the nucleotide diversity of PkTRAMP (π = 0.009) was found to be lower than that of other P. knowlesi genes such as merozoite surface protein 7D (MSP7D) (π = 0.052) (Ahmed and Quan 2019), merozoite surface protein 3 (MSP3) (π = 0.046) (De Silva et al. 2017), and circumsporozoite protein (csp) (π = 0.020) (Chong et al. 2020), but almost similar to other low polymorphism genes such as AMA-1 (π = 0.006) (Ng et al. 2021) and merozoite surface protein 4 (MSP4) (π = 0.007) (Ahmed et al. 2019). The haplotype diversity of PkTRAMP (Hd = 0.999) was similar to other P. knowlesi genes such as AMA-1 (Hd = 1.000) (Ng et al. 2021) and MSP3 (Hd = 0.999) (De Silva et al. 2017).
The identification of high haplotype diversity but low nucleotide diversity in PkTRAMP sequences was similar to those of other P. knowlesi genes such as AMA-1 (Ng et al. 2021), rhoptry-associated protein 1 (RAP-1) (Rawa et al. 2016), and MSP4 (Ahmed et al. 2019). This may suggest that the population may have undergone a recent expansion (Grant and Bowen 1998). Although haplotype diversity was high, the low nucleotide diversity values indicate minor variations between haplotypes. This is supported by the haplotype network analysis, which revealed mostly single or minor differences between haplotypes (Fig. 3).
The haplotype network revealed no distinct geographical separation of PkTRAMP between Peninsular Malaysia and Malaysian Borneo. This is similar to P. knowlesi proteins such as the PkCSP (Chong et al. 2020) and PkMSP7D (Ahmed and Quan 2019). However, some P. knowlesi genetic markers such as PkDBPαII (Fong et al. 2015), PkAMA-1 (Ng et al. 2021), PkCOXI, and PkA-type 18S rRNA (Yusof et al. 2016) showed distinct geographical separation.
The Z-test for natural selection on PkTRAMP indicated that PkTRAMP was under negative (purifying) selection, which is similar to other P. knowlesi genes (Fong et al. 2015; Rawa et al. 2016; Ahmed et al. 2019; Ng et al. 2021). This could possibly be due to functional constraints of the proteins that limit diversity since these genes have been shown to play important roles in the parasite’s invasion into host erythrocyte. Another possible explanation could be that the P. knowlesi samples collected in these studies originated from human infections. Previous studies have shown that P. knowlesi invasion into macaque and human erythrocytes differs. For example, the P. knowlesi Duffy binding protein (DBP) has been shown to be crucial for human erythrocyte invasion but not macaque erythrocyte invasion (Dankwa et al. 2016). Thus, in theory, there would be a greater selection pressure towards certain P. knowlesi proteins in the human host rather than the macaque host, resulting in an increased representation of these haplotypes in the parasite population infecting humans.
Conclusion
The present study revealed low polymorphism of PkTRAMP and an absence of geographical clustering of the protein. In addition, PkTRAMP was found to be undergoing negative selection. Future genetic studies involving larger number of samples and immuno-characterisation of PkTRAMP should be carried out to validate the protein as a vaccine candidate for P. knowlesi malaria.
Data availability
All datasets are presented in the manuscript. Nucleotide sequences generated in the study are deposited into GenBank.
References
Adams JC, Tucker RP (2000) The thrombospondin type 1 repeat (TSR) superfamily: diverse proteins with related roles in neuronal development. Dev Dyn 218:280–299. https://doi.org/10.1002/(SICI)1097-0177(200006)218:2/3C280::AID-DVDY4/3E3.0.CO;2-0
Ahmed MA, Quan FS (2019) Plasmodium knowlesi clinical isolates from Malaysia show extensive diversity and strong differential selection pressure at the merozoite surface protein 7D (MSP7D). Malar J 18:150. https://doi.org/10.1186/s12936-019-2782-2
Ahmed MA, Saif A, Quan FS (2019) Diversity pattern of Plasmodium knowlesi merozoite surface protein 4 (MSP4) in natural population of Malaysia. PLoS ONE 14:e0224743. https://doi.org/10.1371/journal.pone.0224743
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Chin AZ, Maluda MCM, Jelip J, Jeffree MSB, Culleton R, Ahmed K (2020) Malaria elimination in Malaysia and the rising threat of Plasmodium knowlesi. J Physiol Anthropol 39:36. https://doi.org/10.1186/s40101-020-00247-5
Chong ETJ, Neoh JWF, Lau TY, Lim YA, Chai HC, Chua KH, Lee PC (2020) Genetic diversity of circumsporozoite protein in Plasmodium knowlesi isolates from Malaysian Borneo and Peninsular Malaysia. Malar J 19:377. https://doi.org/10.1016/j.cell.2006.02.006
Cowman AF, Crabb BS (2006) Invasion of red blood cells by malaria parasites. Cell 124:755–766. https://doi.org/10.1016/j.cell.2006.02.006
Cox-Singh J, Davis TM, Lee KS, Shamsul SS, Matusop A, Ratnam S, Rahman HA, Conway DJ, Singh B (2008) Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin Infect Dis 46:165–171. https://doi.org/10.1086/524888
Dankwa S, Lim C, Bei AK, Jiang RH, Abshire JR, Patel SD, Goldberg JM, Moreno Y, Kono M, Niles JC et al (2016) Ancient human sialic acid variant restricts an emerging zoonotic malaria parasite. Nat Commun 7:11187. https://doi.org/10.1038/ncomms11187
De Silva JR, Lau YL, Fong MY (2017) Genetic clustering and polymorphism of the merozoite surface protein-3 of Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Parasit Vectors 10:2. https://doi.org/10.1186/s13071-016-1935-1
Fong MY, Rashdi SA, Yusof R, Lau YL (2015) Distinct genetic difference between the Duffy binding protein (PkDBPαII) of Plasmodium knowlesi clinical isolates from North Borneo and Peninsular Malaysia. Malar J 14:91. https://doi.org/10.1186/s12936-015-0610-x
Grant WAS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89:415–426. https://doi.org/10.1093/jhered/89.5.415
Hussin N, Lim YA, Goh PP, William T, Jelip J, Mudin RN (2020) Updates on malaria incidence and profile in Malaysia from 2013 to 2017. Malar J 19:55. https://doi.org/10.1186/s12936-020-3135-x
Imwong M, Tanomsing N, Pukrittayakamee S, Day NP, White NJ, Snounou G (2009) Spurious amplification of a Plasmodium vivax small-subunit RNA gene by use of primers currently used to detect P. knowlesi. J Clin Microbiol 47:4173–4175. https://doi.org/10.1128/JCM.00811-09
Knowles R, Gupta BMD (1932) A study of monkey-malaria and its experimental transmission to man. Ind Med Gaz 67:301–320
Luchavez J, Espino F, Curameng P, Espina R, Bell D, Chiodini P, Nolder D, Sutherland C, Lee KS, Singh B (2008) Human infections with Plasmodium knowlesi, the Philippines. Emerg Infect Dis 14:811–813. https://doi.org/10.3201/eid1405.071407
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426. https://doi.org/10.1093/oxfordjournals.molbev.a040410
Ng OT, Ooi EE, Lee CC, Lee PJ, Ng LC, Pei SW, Tu TM, Loh JP, Leo YS (2008) Naturally acquired human Plasmodium knowlesi infection, Singapore. Emerg Infect Dis 14:814–816. https://doi.org/10.3201/eid1405.070863
Ng YL, Fong MY, Lau YL (2021) Genetic diversity of the full length apical membrane antigen-1 of Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Trop Biomed 38:159–164. https://doi.org/10.47665/tb.38.2.052
Rawa MS, Fong MY, Lau YL (2016) Genetic diversity and natural selection in the rhoptry-associated protein 1 (RAP-1) of recent Plasmodium knowlesi clinical isolates from Malaysia. Malar J 15:62. https://doi.org/10.1186/s12936-016-1127-7
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Siddiqui FA, Dhawan S, Singh S, Singh B, Gupta P, Pandey A, Mohmmed A, Gaur D, Chitnis CE (2013) A thrombospondin structural repeat containing rhoptry protein from Plasmodium falciparum mediates erythrocyte invasion. Cell Microbiol 15:1341–1356. https://doi.org/10.1111/cmi.12118
Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, Thaithong S, Brown KN (1993) High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol 61:315–320. https://doi.org/10.1016/0166-6851(93)90077-B
Thompson J, Cooke RE, Moore S, Anderson LF, Janse CJ, Waters AP (2004) PTRAMP; a conserved Plasmodium thrombospondin-related apical merozoite protein. Mol Biochem Parasitol 134:225–232. https://doi.org/10.1016/j.molbiopara.2003.12.003
Uchime O, Herrera R, Reiter K, Kotova S, Shimp RL Jr, Miura K, Jones D, Lebowitz J, Ambroggio X, Hurt DE et al (2012) Analysis of the conformation and function of the Plasmodium falciparum merozoite proteins MTRAP and PTRAMP. Eukaryot Cell 11:615–625. https://doi.org/10.1128/EC.00039-12
Van den Eede P, Van HN, Van Overmeir C, Vythilingam I, Duc TN, le Hung X, Manh HN, Anné J, D’Alessandro U, Erhart A (2009) Human Plasmodium knowlesi infections in young children in central Vietnam. Malar J 8:249. https://doi.org/10.1186/1475-2875-8-249
WHO (2021) World Health Organisation, World Malaria Report 2020.
Yusof R, Ahmed MA, Jelip J, Ngian HU, Mustakim S, Hussin HM, Fong MY, Mahmud R, Sitam FA, Japning JR et al (2016) Phylogeographic evidence for 2 genetically distinct zoonotic Plasmodium knowlesi parasites, Malaysia. Emerg Infect Dis 22:1371–1380. https://doi.org/10.3201/eid2208.151885
Acknowledgements
We would like to thank the health officials from the State Health Departments and District Health Offices in Pahang, Kelantan, Johor, Kedah, Terengganu, Perak, Negeri Sembilan, Selangor, Federal Territory of Kuala Lumpur, Sabah, and Sarawak for their assistance in sample collection.
Funding
This study was funded by the Ministry of Higher Education Malaysia Long Term Research Grant Scheme (LRGS/1/2018/UM/01/1/1).
Author information
Authors and Affiliations
Contributions
Mun Yik Fong and Yee Ling Lau conceived the idea, design the study, and developed the methodology. Mohd Hafizi Abdul Hamid, Jenarun Jelip, Choo Huck Ooi, Rose Nani Mudin, and Joel Judson Jaimin were involved in the collection and screening of human blood samples for malaria infection. Yee Ling Ng performed the DNA sequencing of the PkTRAMP gene and molecular data analysis. Yee Ling Ng wrote the manuscript, and other authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Ethics approval
Ethical approval for the use of these blood samples was granted by the Medical Research Subcommittee of the Ministry of Health, Malaysia (NMRR-15–67223975).
Consent to participate
Human blood samples used in this study were taken as part of routine (planned) blood sampling for national malaria screening. No additional blood was drawn. No patients were subjected to additional procedures or were required to follow the rules of behaviour. Therefore, regulations on medical research involving human subject do not apply.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interest.
Additional information
Handling Editor: Una Ryan
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ng, Y.L., Lau, Y.L., Hamid, M.H.A. et al. Genetic polymorphism of the thrombospondin-related apical merozoite protein (TRAMP) of Plasmodium knowlesi in Malaysia. Parasitol Res 122, 195–200 (2023). https://doi.org/10.1007/s00436-022-07716-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-022-07716-z