
Abstract
Visceral leishmaniasis (VL) is endemic in Iran and is caused predominantly by Leishmania infantum, but L. tropica is emerging as an important cause. We studied the intra-species population structure of Leishmania spp. causing VL in southwest Iran by sequence analysis of the internal transcribed spacer (ITS) 1 of DNA samples from 29 bone marrow aspiration smears. L. infantum (n = 25) and L. tropica (n = 4) were identified, consisting of 10 and three ITS1 sequence types (STs), respectively. Compared to GenBank ITS1 STs, our L. infantum parasites displayed high heterogeneity but less heterogeneity compared than northwest Iranian isolates. VL affects mostly nomadic populations in southwest Iran, and their mobility may explain partly the L. infantum heterogeneity. The VL causing L. tropica was also genetically heterogeneous but genetically indistinguishable from L. tropica strains causing anthroponotic cutaneous leishmaniasis from southwest Iran.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Leishmaniasis is a complex of neglected tropical diseases, caused by different species of Leishmania, which are present in five continents (Zijlstra 2016). Visceral leishmaniasis (VL) is the most severe form of disease and is potentially fatal if untreated. Globally, the annual VL incidence is approximately 200,000 to 400,000 cases with 20,000 to 40,000 deaths per year (Alvar et al. 2012).
VL causing species vary geographically, Leishmania donovani in South Asia (Bern et al. 2007; Rijal et al. 2010; Singh et al. 2010) and East Africa (Elnaiem 2011; Zijlstra 2016), and Leishmania infantum (L. chagasi) in Central and South America, the Middle East, Mediterranean Basin, Central Asia and China (WHO 2010). VL caused by Leishmania tropica, a species that causes Old World cutaneous leishmaniasis (CL), has been reported sporadically in humans in South Asia and humans and dogs in the Middle East and North Africa (Mebrahtu et al. 1989; Guessous-Idrissi et al. 1997; Alborzi et al. 2006; Hajjaran et al. 2007; Khanra et al. 2012; Hosseininasab et al. 2014).
In Iran, L. infantum is the main cause of VL. However, L. tropica and L. major, the organisms causing anthroponotic and zoonotic cutaneous leishmaniasis (A and ZCL), respectively, may also cause rarely VL with L. tropica being more common than L. major (Karamian et al. 2007; Mohebali 2013). The principal VL foci in Iran are in the northwest (NW) provinces of East Azerbaijan and Ardabil and southwest (SW) provinces of Fars, Bushehr, Kohgiloye and Boyerahmad and Kerman; most VL cases are reported from Fars (Sarkari et al. 2012; Sarkari et al. 2016).
Understanding the population structure and phylogenetic divergence of Leishmania spp. at the intra-species level in time and place could provide way of tracking epidemiological changes in leishmaniasis (Ghatee et al. 2014). Multilocus isoenzyme electrophoresis (MLEE) has long been the gold standard for identifying Leishmania species and intra-species differences (Pratlong et al. 1991). However, because of its lower intra-species discriminatory power and the need for parasite mass cultivation, it has been superseded by other techniques as multilocus sequence typing (MLST), multilocus microsatellite typing (MLMT), polymerase chain reaction restriction fragment length polymorphism (PCR-RFLP) and sequencing of specific genetic markers such as hsp70, ribosomal DNA (rDNA) and the kinetoplastid DNA (kDNA) (Schönian et al. 2008, Schönian et al. 2011; Yuan et al. 2017; Ghatee et al. 2018b). Of these, MLMT and kDNA-RFLP are the most powerful techniques for studying Leishmania populations in intra-species level (Schönian et al. 2011, Schönian et al. 2013). However, challenges remain. MLMT and MLST have been confined in developing countries due to expensive cost. Aside from kDNA, PCR-RFLP of genes has not shown sufficiently high resolution for population structure studies, but kDNA-RFLP is associated with variable DNA yields and low reproducibility of results (Bhattarai et al. 2010; Downing et al. 2011; Ghatee et al. 2014).
The internal transcribed spacers 1 and 2 (ITS1 and ITS2) of the rDNA show high genetic variation and have been used for species typing (Schönian et al. 2003; Parvizi et al. 2008; Yang et al. 2010; de Almeida et al. 2011; Al-Jawabreh et al. 2017) as well as intra-species level phylogenetic and population structure studies of Leishmania species (El Tai et al. 2001; Schönian et al. 2001; Mauricio et al. 2004; Pandey et al. 2007; Parvizi et al. 2008; Talmi-Frank et al. 2010; Raju et al. 2012; Fakhar et al. 2016; Ben Othman et al. 2018) including different studies in Iran (Kuhls et al. 2005; Tashakori et al. 2006; Dabirzadeh et al. 2012; Ghatee et al. 2013a, Ghatee et al. 2014; Karamian et al. 2016).
ITS1 has greater polymorphism than ITS2 and shown greater discrimination of L. donovani and L. tropica in phylogenetic studies in Sudan and Iran, respectively (El Tai et al. 2001; Ghatee et al. 2014). Moreover, ITS1 is more frequently deposited in GenBank, and this allows a greater opportunity for comparing genotypes. There are no data on the genetic population structure of VL causing Leishmania spp. from southern Iran. Therefore, we conducted an ITS1 sequence analysis of VL causing Leishmania spp. in southwest Iran and compared the population structure with those from other regions of Iran.
Material and methods
Study area and patients
Bone marrow aspiration smears from 29 patients with microscopically confirmed kala-azar were obtained from the pathology laboratories of Shahid Faghihi (SF) hospital (n = 26), Shiraz, the capital of Fars Province in SW Iran and Afzalipour hospital (n = 3), Kerman City, the capital of Kerman Province in south-southeast Iran. The SF hospital is a referral centre and receives patients from neighbouring provinces. Patients’ home addresses were retrieved from the hospital records. The VL patients were from Fars, Bushehr, Kohgiloyeh and Boyerahmad Provinces, the kala-azar endemic regions in southwest Iran and two counties in south-southeast province of Kerman in the close proximity of Fars Province. These provinces are also endemic foci of CL. This study was approved by the Ethic Committee of Yasuj University of Medical Sciences (IR.YUMS.1.REC.1395.3), and patients’ record were anonymized and de-identified prior to analysis.
Microscopic examination & DNA extraction
Bone marrow aspiration smears were re-examined microscopically to reconfirm the presence of Leishman bodies and were scratched using sterile scalpels. The sample scrapings were placed in 1.5 ml microtubes containing lysis buffer (Tris 100 mM, EDTA 10 mM, NaCl 100 mM, SDS 1%, Triton X100 2%). Ten micrograms per millilitre of proteinase K was added to the mixture and was incubated at 56 °C for 1 h and then extracted with phenol/chloroform (25:24 v/v) first and then again with chloroform. DNA was precipitated with equal volumes of iso-propanol and one tenth volume of 3 M NaAc. The deposited DNA was washed with 70% ethanol, dried, and suspended in 30 μl of ultrapure water.
Kinetoplastid DNA PCR
To identify the species of the Leishmania isolates, kDNA was amplified using the primers 13Z (5′-ACT GGG GGT TGG GTG TAA AATAG- 3′) and LiR (5′-TCG CAG AAC GCC CCT-3′) (Noyes et al. 1998). The PCR mixture contained 12.5 μl of 2× premix (Ampliqon, Denmark), 20 pmol of each primer, 5 μl of template DNA, and molecular grade water up to 25 μl. Reference strains and negative and inhibition controls were used for each experiment. The PCR process included a pre-denaturing step at 95 °C for 5 min followed by 35 cycles at 94 °C for 45 s, 55 °C for 60 s and 72 °C for 90 s in an Applied Biosystems thermocycler. The PCR products were electrophoresed in 1.2% agarose gel with 0.5 μg/ml ethidium bromide for 90 min at 80 Vin TBE buffer and visualised by a transilluminator. A 100-bp DNA marker was used in each experiment as the standard size. Previously defined isolates of L. major (MRHO/IR/75/ER) and L. infantum (MCAN/IR/07/Moheb-gh) and also L. tropica from a clinical isolate with a laboratory confirmed species finding, preserved in RPMI 1640 culture media, were used as reference strains.
ITS1-rDNA PCR
ITS1-rDNA was amplified using primers LITSR (forward, 5′-CTG GAT CAT TTT CCG ATG-3′) and L5.8S (reverse, 5_-TGA TAC CAC TTA TCG CAC TT-3′), as described by Schönian et al. (2003). The PCR mixture consisted of 25 μl of 2× premix (Ampliqon, Denmark), 20 pmol of each primer, 10 μl (50 ng) of template DNA, and molecular grade water up to 50 μl. Negative and inhibition controls were used for each PCR run. The cycling PCR conditions were 95 °C for 5 min followed by 35 cycles of 94 °C for 30 s, 53 °C for 30 s and 72 °C for 60 s, and a final elongation step at 72 °C for 5 min. The PCR products were electrophoresed on 1.5% agarose gel electrophoresis with 0.5 μg/ml of ethidium bromide for 60 min at 80 V and visualised under the same conditions, as described above.
Sequence analysis
The PCR products of ITS1 DNA were excised from the gel and purified using a gel purification kit (Bioneer, South Korea), according to the manufacturer’s instructions, and were sent to Bioneer Company (South Korea) for sequence analysis using an Applied Biosystems 3730 XL automated DNA sequencer. Sequencing was performed in both directions using by the same primers used for ITS1 amplification. The ITS1 sequences were deposited in the GenBank database under the accession numbers KY964622–KY964634.
Phylogenetic analysis
Species identification, based on the kDNA-PCR, was confirmed by comparison of our ITS1 sequences with those published in the GenBank database using BLAST software (http://www.ncbi.nlm.nih.gov). The obtained sequences were analysed by Geneious Pro 5.5.6 software (http://www.geneious.com, Kearse et al. 2012). Phylogenetic trees were generated by using L. infantum and L. tropica ITS1 sequence types (ST) from this study and those from GenBank, namely L. infantum from Iran and L. tropica from Iran and west Afghanistan (the eastern neighbour of Iran). The maximum likelihood (ML) tree was constructed using MEGA 6 software (Tamura et al. 2013) after trimming all sequences at both ends. Bootstrap values for the ML method were based on 1000 replicates. Figure 1 shows the provinces from where sequence types were obtained.

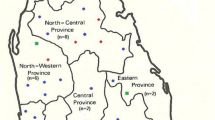
The provinces where ITS1 sequences of L. infantum (a) and L. tropica (b) were obtained and retrieved from GenBank are shown in yellow and orange, respectively. The green circles show provinces that our L. infantum and L. tropica were isolated from in current study. Most isolates were from Fars Province. In map A, Baft County from Kerman Province was shown
Results
The patient’s demographic data are shown in Table 1. Based on the species-specific size, 25 and four samples were identified as L. infantum (680 bp) and L. tropica (750 bp), respectively, by using kDNA PCR. The species identification was confirmed by comparing our ITS1 sequences with sequences that have been deposited in GenBank.
Sequence types—L. infantum
Ten haplotypes were found among the 25 L. infantum ITS1 sequences. ST1 was the most frequent [n = 10 (40%)], followed by ST4 (n = 4), ST2 (n = 3), ST3 (n = 2), and one each for ST5–ST10. All nucleotide variations were found in the first 120 upstream nucleotides, which occurred mostly in the first 60 nucleotides and in the microsatellite sequences, including CC, AA, and TA repeats (Fig. 2). Most similarity was found between ST1 and ST4 (99.6%), ST4 and ST7 (99.2%), and ST4 and ST3 (99.2%), and the least similarity was found between ST10 and ST8 (92.7%), and ST10 and ST6 (93%) (Fig. 3).

Alignment of 10 sequence types obtained from the 25 L. infantum isolates in our study from southwest Iran. Most variations including single nuclear polymorphisms and repeat deletions and insertions were found in the microsatellite sequences

The similarity of different L. infantum sequence types obtained from southwest Iran in this study. The cells with higher similarity are darker in colour
Sequence types—L. tropica
Three STs were found among the four L. tropica parasites. STA was found in two isolates and STB and STC in the remaining two. Greater similarity was present between STA and STB in comparison to STA or B vs. STC. Deletion of CA was found in the region with repeats of C in the upstream part of STB in comparison to STA, but STC showed more differences in upstream and downstream regions that are shown in Fig. 4.

Alignment of three sequence types from visceral leishmaniasis patients from southwest Iran. Sequence type C showed the highest differences in the inter-transcript spacer 1, including insertions, deletions, trans versions, and transition changes
The VL L. tropica STs were compared with ACL haplotypes from previous studies conducted in different parts of Iran. STA was the same as ST4 from the east Iran population. In the second rank, most similarity was found between STA and (i) ST3 and ST5 (99.6%) from east Iran, and (ii) haplotype (Hap) A (99.2%) from southeast (SE) Iran. STC showed the most ST differences compared to ACL L. tropica STs and haplotypes from SW, SE and east Iran (Fig. 5). Table 2 shows the geographical distribution of L. infantum and L. tropica STs in this study; the names of STs and Hap from east and SE are the same as those reported in the original papers (Ghatee et al. 2014; Karamian et al. 2016).

The similarity of three L. tropica sequence types obtained from visceral leishmaniasis patients from southwest Iran compared to GenBank sequence types from patients with anthroponotic cutaneous leishmaniasis due to L. tropica from southwest, southeast, and eastern Iran. ST1–7 (east) (Karamian et al. 2016) Hap A, B, and D (southeast), Hap A and C (southwest) (Ghatee et al. 2014)
Phylogenetic trees
The L. infantum STs from different VL foci in Iran (our data and those from GenBank) were distributed in different positions in the maximum likelihood tree. Clade A-i comprised all STs from SW (Fars, Bushehr, Kohgiloyeh and Boyerahmad Provinces), SE (Kerman Province), central (Alborz, Tehran and Yazd Provinces), west (Lorestan Province) and northeast (Golestan Province) Iran and as well as some STs from northwest (NW) Iran (Ardabil Province). In this clade, STs1, 3–4, 7 and 9–10 from our SW isolates made up a subcluster. Additional sub clusters consisted of (i) all ST sin clade A-i from central, NW and NE Iran together with ST2 and a haplotype (KC570454) from SW Iran (GenBank), and (ii) ST8 and one haplotype from west Iran (Lorestan Province). STs 5 and 6 formed separate branches within clade A-i. Clade B-i consisted of only two STs from NW Iran (Ardabil Province) (Fig. 6).

Unrooted maximum likelihood tree inferred from the internal transcribed spacer 1 sequences of 10 representatives of L. infantum from 25 isolates from southwest Iran and those from other parts of Iran retrieved from GenBank. Only one ITS1 L. infantum from Southwest Iran (KC57454) was previously deposited in GenBank. The numbers above the branches show the bootstrap value for 1000 replicates
The two main L. tropica clades were A-t and B-t. Clade A-t comprised most STs and included all isolates from SW, SE, east Iran and western Afghanistan and some isolates from west (Khuzestan Province), NE (Golestan and Khorasan Provinces) Iran. Within clade A-t, the viscerotropic STA (Kangan County, Bushehr Province, SW and Kerman County, Kerman Province, SE), STB (Shiraz county, Fars Province, SW) and other isolates SW (Fars Province), east (South Khorasan), and central (Alborz and Yazd Provinces) Iran and western Afghanistan (Herat Province) are in similar positions.
Three sub clusters are evident for some L. tropica STs from (i) east Iran and western Afghanistan, (ii) west (Khuzestan Province) and central (Isfahan Province) Iran, and (iii) NE (Khorasan Province), SW (Fars Province) Iran and viscerotropic STC from SW Iran (Darab county, Fars Province). One isolate from SE (Kerman Province) and east (South Khorasan Province) regions formed separate branches in clade A-t. The SW population showed the highest heterogeneity in clade A-t. Our viscerotropic L. tropica isolates were also heterogeneous, but they were not genetically diverged from other L. tropica isolates from southwest Iran. Clade B-t consisted of STs from NE (Razavi Khorasan Province) and west (Kermanshah Province) Iran (Fig. 7).

Unrooted maximum likelihood tree inferred from the internal transcribed spacer 1 sequences of three L. tropica isolates from patients with visceral leishmaniasis from southwest (STA also from southeast) Iran and L. tropica sequences from other regions in Iran and west Afghanistan
Discussion
In current study, we have shown that VL in patients from southern Iran was caused mostly by L. infantum and, in a small number of patients, by L. tropica. Ten L. infantum and three L. tropica STs were identified among the 29 ITS1 sequences.
We found high heterogeneity for L. infantum from SW Iran, but these were mostly grouped together on the phylogenetic tree and included the dominant STs. NW Iranian L. infantum isolates had the highest heterogeneity, and some of these isolates formed a distinct clade (B-i), which showed the most divergence compared to other Iranian L. infantum isolates, including those from the NW in clade A-i. The least heterogeneity was found in L. infantum parasites from Central Iran. The VL-associated L. tropica isolates were also heterogeneous, but they were not genetically distinct from other L. tropica isolates that caused ACL in SW Iran.
The clade B-i, NW Iranian L. infantum isolates (GenBank: EU637915 and EU680963), have previously been detected from Phlebotomus perfilewi, the main vector of L. infantum, and dogs in Ardabil Province of NW Iran (Moshfe et al. 2008; Oshaghi et al. 2009) but, interestingly, are also identical to ITS1 from L. donovani (KT438661-2, KT4386674-9, KT438680-1). Confirmation of L. infantum and differentiation from L. donovani was also shown by cysteine protease B (CBP) gene amplification by Oshaghi et al. (2009), in the previous study. Moreover, L. donovani has also been isolated from P. perfilewi sand flies in NW Iran and Meriones persicus, the Persian jird (a rodent), from several areas of Iran (Mohebali et al. 2004; Oshaghi et al. 2009). Iran has not been known as a focus of L. donovani-associated VL, but these data suggest that Iran could support the transmission of L. donovani.
In the Middle East, L. donovani is confined to foci in Turkey, Cyprus and Iraq (WHO 2010; Gouzelou et al. 2012). In the Cukurova region of SE Turkey, bordering NW Iran, there is a major focus of CL caused by L. donovani/infantum hybrid strains, which are transmitted by P. tobbi, the main sandfly species in Cukurova (Seblova et al. 2015). P. tobbi has also been identified as one of the vectors of L. infantum in NW Iran (Rassi et al. 2012). Based on the results of sequence analysis of the ITS1 and Cbp loci and the epidemiological similarities between NW Iran and SE Turkey, we hypothesise that distinct strains of L. infantum from NW Iran may have evolved from the hybrid strains of L. donovani/L. infantum in neighbouring SW Turkey, and this may explain why they formed their own clade in the phylogenetic tree. More work is needed to confirm our hypothesis.
High heterogeneity was also shown in SW Iranian L. infantum haplotypes and may be explained by population movement. Indeed, VL in SW Iran may have begun because of the immigration of nomadic populations from NW Iran in the sixteenth century (Mazloumi Gavgani et al. 2011). Nomadic populations have the highest VL seropositivity rates in Iran (Mohebali et al. 2006) and their movements, especially in semi-arid regions where sand flies thrive, and the proximity of villages to nomadic travel routes are key factors affecting the distribution of VL in Iran (Ghatee et al. 2013a, b). We hypothesise that the high heterogeneity of SW Iranian L. infantum in clade A-i also may be explained by the recent L. infantum population expansion after a bottleneck effect when parasite strains were imported by infected nomads and their infected shepherd dogs in the previous centuries. Different species of sand fly including Ph. major, Ph. alexanderi and Ph. keshishiani have been reported as vectors of L. infantum in SW Iran (Yaghoobi-Ershadi 2012). Variety of sandfly vector species and strains may have role in heterogeneity of Leishmania parasite regarding hypothesis of parasite-vector co-evolution. Evidences of parasite-vector co-evolution for L. tropica and Ph. sergenti strains, the proven vector of L. tropica, were reported in south Iran (Karamian et al. 2016).
VL occurs sporadically in other regions of Iran (Hamzavi et al. 2012) and is emerging in foci in central Iran (Heidari et al. 2015). The similarity of sequence haplotypes from different regions of central Iran can be explained by the clonal expansion of newly established Leishmania parasites, the presence of few nomadic tribes and similar ecosystems in these foci.
CL due to L. infantum has only recently been identified in SW (Motazedian et al. 2002) and NW Iran (Badirzadeh et al. 2013), but the phylogenetic position of these isolates has not been studied until now.
L. tropica causing VL was first reported in a patient in Kenya (Mebrahtu et al. 1989) and has been followed by other human cases from American soldiers deployed in Iraq and individuals from Iran, India, and Kenya and Afghanistan (Magill et al. 1993; Sacks et al. 1995; Alborzi et al. 2006; Weiss et al. 2009; Khanra et al. 2012). India reports the highest caseload of L. tropica-related VL (Sacks et al. 1995). Canine VL due L. tropica has been reported from Iran and Morocco (Guessous-Idrissi et al. 1997; Hajjaran et al. 2007; Mohebali et al. 2011).
The first case of L. tropica VL came from SW Iran (Alborzi et al. 2006), and other cases have been reported countrywide with varying frequencies, e.g., ranging from 1.4% in 1993/4 to 14% in our study (Alborzi et al. 2006; Mohebali 2013; Hosseininasab et al. 2014). Of our four cases, two were obtained from Shiraz and Kerman City, which are well established endemic foci of L. tropica-related ACL (Ghatee et al. 2014; Izadi et al. 2016), while the other two were from areas where L. tropica has not been reported previously. These limited data hint at trend of increasing L. tropica-related VL and emergence in the new regions in Iran that may be related to population movements or global warming that promotes favourable breeding of sand flies (Ghatee et al. 2018a). More work is needed to establish the trend of L. tropica-related VL to inform future control strategies.
Homogeneity of SE and most east and central Iranian and western Afghanistan and the genetic heterogeneity of northeast and SW Iranian ACL-associated L. tropica have been described previously (Ghatee et al. 2014; Fakhar et al. 2016; Karamian et al. 2016). Viscerotropic haplotypes, STA and STB, are in the same position on the phylogenetic tree as the SW ACL isolates and that the STC haplotype clustered with other SW ACL isolates. Overall, our three L. tropica haplotypes from viscerotropic cases could not diverge from ACL isolates from SW Iran. Indian VL-associated L. tropica isolates were also not discerned from the Indian CL-associated subpopulation (Krayter et al. 2014). The visceralisation of L. tropica may be related partially to reduced host immunity, e.g., HIV positivity (Jafari et al. 2010) and/or increased pathogenicity of strains (Krayter et al. 2014). HIV and HIV/VL co-infection rate seems to increase in Iran where HIV/VL occurrence was reported up to 18% of HIV-positive patients (Shafiei et al. 2014; Gökengin et al. 2016) that may be led to higher incidence of the visceralisation of cutaneous or asymptomatic Leishmania infections.
This is an area where greater understanding of L. tropica viscerotropism is needed.
Conclusion
We found 10 heterogeneous L. infantum and three heterogeneous L. tropica STs from patients with VL from SW Iran. The high heterogeneity of SW and NW L. infantum may be due to within-country population movements. Central Iran has the least heterogeneity where only small VL foci exist. L. tropica causing VL from SW Iran was heterogonous but was not distinct genetically from ACL-related L. tropica from SW Iran. Mechanisms underlying the visceralisation of L. tropica remain unknown. More research is needed to understand the dynamics of VL-related L. infantum and L. tropica and to understand the adaptation of L. tropica to visceral disease.
References
Alborzi A, Rasouli M, Shamsizadeh A (2006) Leishmania tropica–isolated patient with visceral leishmaniasis in southern Iran. Am J Trop Med Hyg 74(2):306–307
Al-Jawabreh A, Dumaidi K, Ereqat S, Al-Jawabreh H, Nasereddin A, Azmi K, Barghuthy F, Sawalha S, Salah I, Abdeen Z (2017) Molecular epidemiology of human cutaneous leishmaniasis in Jericho and its vicinity in Palestine from 1994 to 2015. Infect Genet Evol 50:95–101. https://doi.org/10.1016/j.meegid.2016.06.007
Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M, Leishmaniasis Control Team WHO (2012) Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7(5):e35671. https://doi.org/10.1371/journal.pone.0035671
Badirzadeh A, Mohebali M, Ghasemian M, Amini H, Zarei Z, Akhoundi B, Hajjaran H, Emdadi D, Molaiei S, Kusha A, Alizadeh S (2013) Cutaneous and post kala-azar dermal leishmaniasis caused by Leishmania infantum in endemic areas of visceral leishmaniasis, northwestern Iran 2002–2011: a case series. Pathog Glob Health 107(4):194–197. https://doi.org/10.1179/2047773213Y.0000000097
Bern C, Haque R, Chowdhury R, Ali M, Kurkjian KM, Vaz L, Amann J, Wahed M, Wagatsuma Y, Breiman RF (2007) The epidemiology of visceral leishmaniasis and asymptomatic leishmanial infection in a highly endemic Bangladeshi village. Am J Trop Med Hyg 76(5):909–914
Bhattarai N, Dujardin J, Rijal S, De Doncker S, Boelaert M, Van der Auwera G (2010) Development and evaluation of different PCR-based typing methods for discrimination of Leishmania donovani isolates from Nepal. Parasitology 137(6):947–957. https://doi.org/10.1017/S0031182009991752
Dabirzadeh M, Mirmohammadsadeghi H, Baghaie M, Hejazi H (2012) Genetic polymorphism of Leishmania major in two hyper endemic regions of Iran revealed by PPIP-PCR and ITS-RFLP. Arch Iran Med 15(3):151–156 https://doi.org/012153/AIM.009
de Almeida ME, Steurer FJ, Koru O, Herwaldt BL, Pieniazek NJ, da Silva AJ (2011) Identification of Leishmania spp. by molecular amplification and DNA sequencing analysis of a fragment of the rRNA internal transcribed spacer 2 (ITS2). J Clin Microbiol 49(9):3143–3149. https://doi.org/10.1128/JCM.01177-11
Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, de Doncker S, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Schönian G, Stark O, Sundar S, Vanaerschot M, Hertz-Fowler C, Dujardin JC, Berriman M (2011) Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res 21(12):2143–2156. https://doi.org/10.1101/gr.123430.111
El Tai NO, El Fari M, Mauricio I, Miles MA, Oskam L, El Safi SH, Presber WH, Schönian G (2001) Leishmania donovani: intraspecific polymorphisms of Sudanese isolates revealed by PCR-based analyses and DNA sequencing. Exp Parasitol 97(1):35–44. https://doi.org/10.1006/expr.2001.4592
Elnaiem DEA (2011) Ecology and control of the sand fly vectors of Leishmania donovani in East Africa, with special emphasis on Phlebotomus orientalis. J Vector Ecol 36:S23–S31. https://doi.org/10.1111/j.1948-7134.2011.00109.x
Fakhar M, Ghohe HP, Rasooli SA, Karamian M, Mohib AS, Hezarjaribi HZ, Pagheh AS, Ghatee MA (2016) Genetic diversity of Leishmania tropica strains isolated from clinical forms of cutaneous leishmaniasis in rural districts of Herat province, western Afghanistan, based on ITS1-rDNA. Infect Genet Evol 41:120–127. https://doi.org/10.1016/j.meegid.2016.03.031
Ghatee M, Sharifi I, Mirhendi H, Kanannejad Z, Hatam G (2013a) Investigation of double-band electrophoretic pattern of ITSrDNA region in Iranian isolates of Leishmania tropica. Iran J Parasitol 8(2):264–272
Ghatee MA, Sharifi I, Haghdoost AA, Kanannejad Z, Taabody Z, Hatam G, Abdollahipanah A (2013b) Spatial correlations of population and ecological factors with distribution of visceral leishmaniasis cases in southwestern Iran. J Vector Borne Dis 50(3):179–187
Ghatee MA, Sharifi I, Kuhls K, Kanannejad Z, Harandi MF, de Almeida ME, Hatam G, Mirhendi H (2014) Heterogeneity of the internal transcribed spacer region in Leishmania tropica isolates from southern Iran. Exp Parasitol 144:44–51. https://doi.org/10.1016/j.exppara.2014.06.003
Ghatee MA, Haghdoost AA, Kooreshnia F, Kanannejad Z, Parisaie Z, Karamian M, Moshfe A (2018a) Role of environmental, climatic risk factors and livestock animals on the occurrence of cutaneous leishmaniasis in newly emerging focus in Iran. J Infect Public Health 11(3):425–433. https://doi.org/10.1016/j.jiph.2017.12.004
Ghatee MA, Mirhendi H, Marashifard M, Kanannejad Z, Taylor WR, Sharifi I (2018b) Population Structure ofLeishmania tropicaCausing Anthroponotic Cutaneous Leishmaniasis in Southern Iran by PCR-RFLP of Kinetoplastid DNA. Biomed Res Int 2018:6049198–6049111. https://doi.org/10.1155/2018/6049198
Gökengin D, Doroudi F, Tohme J, Collins B, Madani N (2016) HIV/AIDS: trends in the Middle East and North Africa region. Int J Infect Dis 44:66–73. https://doi.org/10.1016/j.ijid.2015.11.008
Gouzelou E, Haralambous C, Amro A, Mentis A, Pratlong F, Dedet JP, Votypka J, Volf P, Toz SO, Kuhls K (2012) Multilocus microsatellite typing (MLMT) of strains from Turkey and Cyprus reveals a novel monophyletic L. donovanisensulato group. PLoS Negl Trop Dis 6(2):e1507. https://doi.org/10.1371/journal.pntd.0001507
Guessous-Idrissi N, Berrag B, Riyad M, Sahibi H, Bichichi M, Rhalem A (1997) Short report: Leishmania tropica: etiologic agent of a case of canine visceral leishmaniasis in northern Morocco. Am J Trop Med Hyg 57(2):172–173
Hajjaran H, Mohebali M, Zarei Z, Edrissian G (2007) Leishmania tropica: another etiological agent of canine visceral Leishmaniasis in Iran. Iran J Public Health 36:85–88
Hamzavi Y, Hamzeh B, Mohebali M, Akhoundi B, Ajhang K, Khademi N, Ghadiri K, Bashiri H, Pajhouhan M (2012) Human visceral leishmaniasis in Kermanshah province, western Iran, during 2011-2012. Iran J Parasitol 7(4):49–56
Heidari A, Mohebali M, Kabir K, Barati H, Soultani Y, Keshavarz H, Akhoundi B, Hajjaran H, Reisi H (2015) Visceral leishmaniasis in rural areas of Alborz province of Iran and implication to health policy. Korean J Parasitol 53(4):379–383. https://doi.org/10.3347/kjp.2015.53.4.379
Hosseininasab A, Sharifi I, Mohammad Hossein D, Zarean M, Dadkhah M (2014) Causes of pediatric visceral leishmaniasis in southeastern Iran. Iran J Parasitol 9(4):584–587
Izadi S, Mirhendi H, Jalalizand N, Khodadadi H, Mohebali M, Nekoeian S, Jamshidi A, Ghatee MA (2016) Molecular epidemiological survey of cutaneous Leishmaniasis in two highly endemic metropolises of Iran, application of FTA cards for DNA extraction from Giemsa-stained slides. Jundishapur J Microbiol 9(2):e32885 https://doi.org/10.5812/jjm.32885
Jafari S, Hajiabdolbaghi M, Mohebali M, Hajjaran H, Hashemian H (2010) Disseminated leishmaniasis caused by Leishmania tropica in HIV-positive patients in the Islamic Republic of Iran. East Mediterr Health J 16(3):340–343
Karamian M, Motazedian MH, Mehrabani D, Gholami K (2007) Leishmania major infection in a patient with visceral leishmaniasis: treatment with amphotericin B. Parasitol Res 101(5):1431–1434. https://doi.org/10.1007/s00436-007-0649-x
Karamian M, Kuhls K, Hemmati M, Ghatee MA (2016) Phylogenetic structure of Leishmania tropica in the new endemic focus Birjand in East Iran in comparison to other Iranian endemic regions. Acta Trop 158:68–76. https://doi.org/10.1016/j.actatropica.2016.02.010
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Khanra S, Datta S, Mondal D, Saha P, Bandopadhyay SK, Roy S, Manna M (2012) RFLPs of ITS, ITS1 and hsp70 amplicons and sequencing of ITS1 of recent clinical isolates of kala-azar from India and Bangladesh confirms the association of L. tropica with the disease. Acta Trop 124(3):229–234. https://doi.org/10.1016/j.actatropica.2012.08.017
Krayter L, Bumb RA, Azmi K, Wuttke J, Malik MD, Schnur LF, Salotra P, Schönian G (2014) Multilocus microsatellite typing reveals a genetic relationship but, also, genetic differences between Indian strains of Leishmania tropica causing cutaneous leishmaniasis and those causing visceral leishmaniasis. Parasit Vectors 7:123. https://doi.org/10.1186/1756-3305-7-123
Kuhls K, Mauricio IL, Pratlong F, Presber W, Schönian G (2005) Analysis of ribosomal DNA internal transcribed spacer sequences of the Leishmania donovani complex. Microbes Infect 7(11–12):1224–1234. https://doi.org/10.1016/j.micinf.2005.04.009
Magill AJ, Grogl M, Gasser RA Jr, Sun W, Oster CN (1993) Visceral infection caused by Leishmania tropica in veterans of operation desert storm. N Engl J Med 328(19):1383–1387 https://doi.org/10.1056/NEJM199305133281904
Mauricio I, Stothard J, Miles M (2004) Leishmania donovani complex: genotyping with the ribosomal internal transcribed spacer and the mini-exon. Parasitology 128(Pt3):263–267
Mazloumi Gavgani AS, Maleki Ravasan N, Mazloumi Gavgani F (2011) Comparison of nomadic and non-nomadic lifestyles in transmission of visceral Leishmaniasis. J Gorgan Uni Med Sci 13:94–100 [article in Persian]
Mebrahtu Y, Lawyer P, Githure J, Were JB, Muigai R, Hendricks L, Leeuwenburg J, Koech D, Roberts C (1989) Visceral leishmaniasis unresponsive to pentostam caused by Leishmania tropica in Kenya. Am J Trop Med Hyg 41(3):289–294
Mohebali M (2013) Visceral leishmaniasis in Iran: review of the epidemiological and clinical features. Iran J Parasitol 8(3):348–358
Mohebali M, Javadian E, Yaghoobi Ershadi M, Akhavan A, Hajjaran H, Abaei M (2004) Characterization of Leishmania infection in rodents from endemic areas of the Islamic Republic of Iran. East Mediterr Health J 10(4–5):591–599
Mohebali M, Edrissian G, Nadim A, Hajjaran H, Akhoundi B, Hooshmand B, Zarei Z, Arshi S, Mirsamadi N, Naeini KM (2006) Application of direct agglutination test (DAT) for the diagnosis and seroepide-miological studies of visceral leishmaniasis in Iran. Iran J Parasitol 1:15–25
Mohebali M, Malmasi A, Hajjaran H, Jamshidi S, Akhoundi B, Rezaei M, Janitabar S, Zarei H, Charehdar S (2011) Disseminated leishmaniasis caused by Leishmania tropica in a puppy from Karaj, Central Iran. Iran J Parasitol 6(2):69–73
Moshfe A, Mohebali M, Edrissian G, Zarei Z, Akhoundi B, Kazemi B, Jamshidi S, Mahmoodi M (2008) Seroepidemiological study on canine visceral Leishmaniasis in Meshkin-Shahr district, Ardabil province, northwest of Iran during 2006-2007. Iran J Parasitol 3:1–10
Motazedian H, Noamanpoor B, Ardehali S (2002) Characterization of Leishmania parasites isolated from provinces of the Islamic Republic of Iran. East Med Health J 8(2–3):338–344
Noyes HA, Reyburn H, Bailey W, Smith D (1998) A nested-PCR-schizodeme method for identifying Leishmania kinetoplastminicircle classes directly from clinical samples and its application to the study of epidemiology of Leishmania tropica in Pakistan. J Clin Microbiol 36(10):2877–2881
Oshaghi MA, Ravasan NM, Hide M, Javadian EA, Rassi Y, Sadraei J, Mohebali M, Sedaghat MM, Hajjaran H, Zarei Z (2009) Phlebotomus perfiliewitranscaucasicus is circulating both Leishmania donovani and L. infantum in Northwest Iran. Exp Parasitol 123(3):218–225. https://doi.org/10.1016/j.exppara.2009.07.004
Othman SB, Ghawar W, Chaouch M, Ayari C, Chemkhi J, Cancino-Faure B, Tomás-Pérez M, Alcover MM, Riera C, Salah AB, Fisa R (2018) First detection of Leishmania DNA in Psammomysobesus and Psammomysvexillaris: their potential involvement in the epidemiology of leishmaniasis in Tunisia. Infect Genet Evol 59:7–15. https://doi.org/10.1016/j.meegid.2018.01.013
Pandey K, Yanagi T, Pandey BD, Mallik AK, Sherchand JB, Kanbara H (2007) Characterization of Leishmania isolates from Nepalese patients with visceral leishmaniasis. Parasitol Res 100 (6):1361–1369
Parvizi P, Moradi G, Akbari G, Farahmand M, Ready PD, Piazak N, Assmar M, Amirkhani A (2008) PCR detection and sequencing of parasite ITS-rDNA gene from reservoirs host of zoonotic cutaneous leishmaniasis in Central Iran. Parasitol Res 103(6):1273–1278. https://doi.org/10.1007/s00436-008-1124-z
Pratlong F, Rioux J, Dereure J, Mahjour J, Gallego M, Guilvard E, Lanotte G, Perieres J, Martini A, Saddiki A (1991) Leishmania tropica in Morocco. IV—intrafocal enzyme diversity. Ann Parasitol Hum Comp 66(3):100–104. https://doi.org/10.1051/parasite/1991663100
Raju BS, Gurumurthy S, Kuhls K, Bhandari V, Schnonian G, Salotra P (2012) Genetic typing reveals monomorphism between antimony sensitive and resistant Leishmania donovani isolates from visceral leishmaniasis or post kala-azar dermal leishmaniasis cases in India. Parasitol Res 111(4):1559–1568. https://doi.org/10.1007/s00436-012-2996-5
Rassi Y, Dehkordi AS, Oshaghi MA, Abai MR, Mohtarami F, Enayati A, Zarei Z, Javadian E (2012) First report on natural infection of the Phlebotomus tobbi by Leishmania infantum in northwestern Iran. Exp Parasitol 131(3):344–349. https://doi.org/10.1016/j.exppara.2012.04.020
Rijal S, Uranw S, Chappuis F, Picado A, Khanal B, Paudel IS, Andersen EW, Meheus F, Ostyn B, Das ML (2010) Epidemiology of Leishmania donovani infection in high-transmission foci in Nepal. Tropical Med Int Health 15:21–28. https://doi.org/10.1111/j.1365-3156.2010.02518.x
Sacks D, Kenney R, Neva F, Kreutzer R, Jaffe C, Gupta A, Sharma M, Sinha S, Saran R (1995) Indian kala-azar caused by Leishmania tropica. Lancet 345(8955):959–961
Sarkari B, Hatam G, Ghatee M (2012) Epidemiological features of visceral leishmaniasis in Fars Province, Southern Iran. Iran J Publi Health 41:94–99
Sarkari B, Naraki T, Ghatee MA, Abdolahi Khabisi S, Davami MH (2016) Visceral Leishmaniasis in Southwestern Iran: a retrospective clinico-hematological analysis of 380 consecutive hospitalized cases (1999–2014). PLoS ONE 11(3): e0150406. https://doi.org/10.1371/journal.pone.0150406
Schönian G, Schnur L, El Fari M, Oskam L, Kolesnikov AA, Sokolowska-Köhler W, Presber W (2001) Genetic heterogeneity in the species Leishmania tropica revealed by different PCR-based methods. Trans R Soc Trop Med Hyg 95(2):217–224
Schönian G, Nasereddin A, Dinse N, Schweynoch C, Schallig HD, Presber W, Jaffe CL (2003) PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis 47(1):349–358
Schönian G, Mauricio I, Gramiccia M, Cañavate C, Boelaert M, Dujardin JC (2008) Leishmaniases in the Mediterranean in the era of molecular epidemiology. Trends Parasitol 24(3):135–142. https://doi.org/10.1016/j.pt.2007.12.006
Schönian G, Kuhls K, Mauricio I (2011) Molecular approaches for a better understanding of the epidemiology and population genetics of Leishmania. Parasitology 138(4):405–425. https://doi.org/10.1017/S0031182010001538
Schönian G, Cupolillo E, Mauricio I (2013) Molecular evolution and phylogeny of Leishmania. In: Ponte-Sucre A, DiazMaritza E, Padrón-Nieves M (eds) Drug resistance in Leishmania parasites. Springer, Vienna, pp 15–44
Seblova V, Myskova J, Hlavacova J, Votypka J, Antoniou M, Volf P (2015) Natural hybrid of Leishmania infantum/L. donovani: development in Phlebotomus tobbi, P. perniciosus and Lutzomyia longipalpis and comparison with non-hybrid strains differing in tissue tropism. Parasit Vectors 8:605. https://doi.org/10.1186/s13071-015-1217-3
Shafiei R, Mohebali M, Akhoundi B, Galian MS, Kalantar F, Ashkan S, Fata A, Farash BR, Ghasemian M (2014) Emergence of co-infection of visceral leishmaniasis in HIV-positive patients in Northeast Iran: a preliminary study. Travel Med Infect Dis 12(2):173–178. https://doi.org/10.1016/j.tmaid.2013.09.001
Singh SP, Picado A, Boelaert M, Gidwani K, Andersen EW, Ostyn B, Meheus F, Rai M, Chappuis F, Davies C (2010) The epidemiology of Leishmania donovani infection in high transmission foci in India. Tropical Med Int Health 15:12–20. https://doi.org/10.1111/j.1365-3156.2010.02519.x
Talmi-Frank D, Jaffe CL, Nasereddin A, Warburg A, King R, Svobodova M, Peleg O, Baneth G (2010) Leishmania tropica in rock hyraxes (Procaviacapensis) in a focus of human cutaneous leishmaniasis. Am J Trop Med Hyg 82(5):814–818. https://doi.org/10.4269/ajtmh.2010.09-0513
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729. https://doi.org/10.1093/molbev/mst197
Tashakori M, Kuhls K, Al-Jawabreh A, Mauricio IL, Schönian G, Farajnia S, Alimohammadian MH (2006) Leishmania major: genetic heterogeneity of Iranian isolates by single-strand conformation polymorphism and sequence analysis of ribosomal DNA internal transcribed spacer. Acta Trop 98(1):52–58. https://doi.org/10.1016/j.actatropica.2006.01.010
Weiss F, Vogenthaler N, Franco-Paredes C, Parker SR (2009) Leishmania tropica–induced cutaneous and presumptive concomitant viscerotropic Leishmaniasis with prolonged incubation. Arch Dermatol 145(9):1023–1026. https://doi.org/10.1001/archdermatol.2009.181
World Health Organization, Control of the leishmaniases: report of a meeting of the WHO expert committee on the control of leishmaniases. Control of the leishmaniases: report of a meeting of the WHO expert committee on the control of leishmaniases. 2010. World Health Organization
Yaghoobi-Ershadi M (2012) Phlebotomine sand flies (Diptera: Psychodidae) in Iran and their role on Leishmania Transmission. J Arthropod Borne Dis 6(1):1–17
Yang BB, Guo XG, Hu XS, Zhang JG, Liao L, Chen DL, Chen JP (2010) Species discrimination and phylogenetic inference of 17 Chinese Leishmania isolates based on internal transcribed spacer 1 (ITS1) sequences. Parasitol Res 107(5):1049–1065. https://doi.org/10.1007/s00436-010-1969-9
Yuan D, Qin H, Zhang J, Liao L, Chen Q, Chen D, Chen J (2017) Phylogenetic analysis of HSP70 and cyt b gene sequences for Chinese Leishmania isolates and ultrastructural characteristics of Chinese Leishmania sp. Parasitol Res 116(2):693–702. https://doi.org/10.1007/s00436-016-5335-4
Zijlstra EE (2016) Visceral leishmaniasis: a forgotten epidemic. Arch Dis Child 101(6):561–567. https://doi.org/10.1136/archdischild-2015-309302
Funding
This study was financially supported by the Vice-Chancellor of Yasuj University of Medical Sciences (project no. P-23-2-263).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ghatee, M.A., Mirhendi, H., Karamian, M. et al. Population structures of Leishmania infantum and Leishmania tropica the causative agents of kala-azar in Southwest Iran. Parasitol Res 117, 3447–3458 (2018). https://doi.org/10.1007/s00436-018-6041-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-018-6041-1