
Abstract
Angiostrongylus cantonensis is an important zoonotic nematode. It is the causative agent of eosinophilic meningitis and eosinophilic meningoencephalitis in humans. However, information of this parasite at the genomic level is very limited. In the present study, the transcriptomic profiles of the fifth-stage larvae (L5) of A. cantonensis were investigated by next-generation sequencing (NGS). In the NGS database established from the larvae isolated from the brain of Sprague–Dawley rats, 31,487 unique genes with a mean length of 617 nucleotides were assembled. These genes were found to have a 46.08 % significant similarity to Caenorhabditis elegans by BLASTx. They were then compared with the expressed sequence tags of 18 other nematodes, and significant matches of 36.09–59.12 % were found. Among these genes, 3,338 were found to participate in 124 Kyoto Encyclopedia of Genes and Genomes pathways. These pathways included 1,514 metabolisms, 846 genetic information processing, 358 environmental information processing, 264 cellular processes, and 91 organismal systems. Analysis of 30,816 sequences with the gene ontology database indicated that their annotations included 5,656 biological processes (3,364 cellular processes, 3,061 developmental processes, and 3,191 multicellular organismal processes), 7,218 molecular functions (4,597 binding and 3,084 catalytic activities), and 4,719 cellular components (4,459 cell parts and 4,466 cells). Moreover, stress-related genes (112 heat stress and 33 oxidation stress) and genes for proteases (159) were not uncommon. This study is the first NGS-based study to set up a transcriptomic database of A. cantonensis L5. The results provide new insights into the survival, development, and host–parasite interactions of this blood-feeding nematode.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Next-generation sequencing (NGS) is an important tool in transcriptomic studies. It greatly decreases the time and effort previously required for DNA sequencing by enabling massively sequencing reactions to define millions or billions base positions and allows identification of targeted genes differentially expressed in different tissues, cells, stages, or genders, during development, or on activation of immune responses. In addition, it is able to obtain complete nuclear and organellar genome sequences simultaneously (Shendure and Ji 2008). NGS technologies such as Roche 454 (Margulies et al. 2005), ABI-SOLiD (Pandey et al. 2008), Solexa (Illumina) (Bentley et al. 2008), and Helicos (Harris et al. 2008) change the way we discover and define parasite transcriptomes and genomes (Droege and Hill 2008; Jex et al. 2010). These sequencing techniques are also involved in the development of enhanced computational methods for the preprocessing, assembly, and annotation of sequence data (Nagaraj et al. 2007a, b). Investigations of the transcriptome by different approaches (Ranganathan et al. 2009; Nisbet et al. 2008) lead to a better understanding of the biochemical and molecular processes involved in the development, reproduction, and parasite–host interactions in a number of parasitic nematodes such as Haemonchus contortus (Campbell et al. 2008; Cantacessi et al. 2010), Ascaris suum (Huang et al. 2008; Cantacessi et al. 2009), Radopholus similis (Jacob et al. 2008), Schistosoma mansoni (Farias et al. 2011), Schistosoma japonicum (Peng et al. 2003), Leishmania donovani (Li et al. 2008), Eimeria brunette (Aarthi et al. 2011), and Clonorchis sinensis (Lee et al. 2003; Cho et al. 2006, 2008).
Angiostrongylus cantonensis, the rat lungworm, is an important zoonotic nematode. This parasite was discovered in the lung of Rattus rattus and Rattus norvegicus in Guangdong in 1933 (Chen 1933). Human is a nonpermissive host and acquires the infection by eating raw or undercooked intermediate hosts (snails or slugs) or paratenic host (frogs) contaminated with the third-stage larvae (L3) (Alicata 1965, 1988). Although Southeast Asia and the Pacific Islands have been considered to be the main endemic regions, more than 2,800 cases have been recorded in 31 countries up to 2010 (Wang et al. 2008, 2012). After penetrating into the human host, the third-stage larvae migrate to the central nervous system via the bloodstream and develop into fifth-stage larvae (L5) after molting twice. At this site, they cause eosinophilic meningitis and eosinophilic meningoencephalitis. Patients have an insidious or sudden onset of excruciating headache, neck stiffness, nausea, vomiting, and paraesthesia, although fever, cranial nerve palsies, seizures, paralysis, and lethargy are less common. Majority of the patients having cerebral angiostrongyliasis usually have a self-limited course and recover without sequelae. However, fatal outcomes have been reported in severe cases (Punyagupta et al. 1975).
We have submitted 1,226 expressed sequence tags (ESTs) of L5 to the National Center for Biotechnology Information (NCBI) dbEST in 2005. These sequences and some additional ones were analyzed and found to encode proteins participating in metabolism, cellular development, immune evasion, and host–parasite interactions (Xu et al. 2009) and were grouped into 13 categories (Fang et al. 2010). After clustering 1,496 ESTs were generated from a cDNA library of L3 into 161 contigs and 757 singletons, 54.5 % of which were found to have a significant sequence homology with known proteins. Among the identified abundant expressive sequences were cathepsin B-like cysteine protease 1 and 2, metalloprotease I, metalloprotease 1 precursor, and extracellular superoxide dismutase (Chang et al. 2011). Since the information obtained by these transcriptomic studies is very limited, the application of the NGS technologies may provide a global study on the gene expressions of A. cantonensis in the central nervous system.
In the present study, we employed a NGS-based approach to set up a transcriptomic database of A. cantonensis L5. The CLC bio software was used to assemble the newly generated sequences and bioinformatic techniques to predict the key Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, gene ontology (GO) annotations, and groups of molecules involved in the fundamental metabolic pathways of the nematode in this pathogenic stage. In addition, comparative analyses of the dataset predicted a range of proteins that are conserved among nematodes.
This study is the first NGS investigation to explore the gene expression of A. cantonensis in a comprehensive manner. The findings provide an invaluable resource to underpin future efforts toward developing new approaches for the intervention against and control of angiostrongyliasis. Understanding the molecular pathways linked to parasite survival in the environment, development, reproduction in the vertebrate host, and host–parasite interactions may provide a guide to the investigations of these pathways and facilitate the identification of new drug targets. Since the genome of A. cantonensis has not been sequenced, the establishment of a complete gene expression database is very important for understanding the infection and pathogenicity mechanism of this parasite.
Materials and methods
Establishment and maintenance of A. cantonensis in laboratory
The life cycle of A. cantonensis was established by isolating more than 10,000 of L3 from an Achatina fulica snail collected in Neihu, Taipei in 1985. After infecting these larvae to Sprague–Dawley (SD) rats, the first-stage larvae (L1) were recovered from the rat feces on day 50 postinfection and fed to Biomphalaria glabrata snails. To isolate L3, the infected B. glabrata snails were killed and the tissues were sonicated with an organization homogenizer (Cole-Parmer Instrument Co., USA) and then digested with artificial gastric juice (0.6 % (w/v) pepsin, pH 2–3) at 37 °C for 45 min on day 20 after infection. By esophageal perfusion, SD rats were infected with the isolated L3. After 42–46 days, L1 were harvested from the rat feces (Wang et al. 1989).
Collection of A. cantonensis L5
After collection from infected B. glabrata, L3 were fed to SD rats by esophageal perfusion. L5 (Fig. 1) were then isolated and collected from rat brains on day 21 postinfection (Wang et al. 1989).

Fifth-stage larvae of A. cantonensis collected from SD rats on day 21 postinfection. Differentiation of stage and gender was based on the sexual organs. In male, the bursa was opened (a head, b tail). The vagina in female was very clear (c head, d tail). Male worms were about 9.69 mm and females were about 11.48 mm in length
RNA extraction
The L5 harvested from rat brains were washed with normal saline, PBS, and ddH2O for three times. The larvae were then disrupted and homogenized in a TRIzol reagent (Molecular Research Center, USA). Chloroform, 75 % (v/v) alcohol (in DEPC-H2O), and isopropanol were used to purify and precipitate RNA samples (Chang et al. 2011).
Next-generation sequencing
Gene expressions in A. cantonensis L5 were identified by direct high-throughput sequencing using the next-generation sequencer Solexa (Illumina, USA). Long RNA sequences were first converted into a library of cDNA fragments, and adaptors were subsequently added to each cDNA fragment. This new sequencing technology (Solexa) relies on the attachment of small DNA fragments to a platform, optically transparent surface, and solid-phase amplification to create an ultrahigh-density sequencing flow cell with >10 million clusters, each containing ∼1,000 copies of template/cm2. These templates were sequenced by a robust four-color DNA sequencing-by-synthesis technology that employs reversible terminators with removable fluorescence. The high-sensitivity fluorescence was then detected by laser excitation. The typical output from a single reaction was approximately 2 GB which contains 20–40 million reads. The Solexa sequence reads were assembled using the CLC bio software. Before assembly, the obtained reads were preprocessed by masking the polyA tails and removing adapters. This generated datasets were analyzed by the Chang Gung Bioinformatics Center.
BLAST homology searches and sequence annotations
Bioinformatic analyses were conducted using FastAnnotator, an efficient transcript annotation web tool (http://fastannotator.cgu.edu.tw/) (Chen et al. 2012). The Caenorhabditis elegans genome database and EST sequences of 18 parasitic or nonparasitic nematodes were obtained from NCBI (http://www.ncbi.nlm.nih.gov/). BLASTx homology searches were performed to compare the proteins inferred from the transcriptomes of A. cantonensis L5 with those predicted from transcriptomic data of C. elegans and the 18 nematodes with an E value cutoff at 1 × 10−5. Identification of GO annotations was carried out using Blast2GO (B2G4Pipe) (Götz et al. 2008). KEGG pathway analysis was performed through the KEGG database (http://www.genome.jp/kegg/).
Results
Characterization of the transcriptome
A total of 15,399,691 short-insert Illumina reads were generated for A. cantonensis L5 with a mean length of 77.51 nucleotides. After removal of adapters, these short-insert Illumina reads were de novo assembled by the CLC bio software (http://www.clcbio.com/index.php?id=1109). The assembly procedure resulted in 31,487 contigs with a mean length of 617 nucleotides (range 128–18,870) and a G+C content of 43.73 %. The total read count was 13,483,766 nucleotides, and the total contig length was 19,440,921 (Table 1).
Sequence homology
By BLAST analysis, 14,509 (46.08 %) of the 31,487 NGS transcriptome contigs from A. cantonensis L5 were found to have significant matches sequences from the C. elegans genome database (E value ≤1 × 10–5) (Table 1). Proteins inferred from the transcriptomes were compared with those predicted from transcriptomic data of 18 nematode ESTs from the NCBI database. Significant matches (36.09–59.12 %) were found with Ancylostoma caninum, Ancylostoma ceylanicum, A. suum, Brugia malayi, Caenorhabditis brenneri, Caenorhabditis remanei, H. contortus, Necator americanus, Nippostrongylus brasiliensis, Onchocerca volvulus, Ostertagia ostertagi, Panagrolaimus superbus, Parastrongyloides trichosuri, Strongyloides ratti, Strongyloides stercoralis, Toxocara canis, Trichinella spiralis, and Trichuris muris (E value ≤1 × 10−5) (Table 2).
Functional analysis
A total of 3,338 sequences were mapped to 124 KEGG pathways (Table 3). A significant proportion of amino acid sequences were associated with (1) metabolic pathways (1,514 sequences) including carbohydrate metabolism, energy metabolism, amino acid metabolism, lipid metabolism, nucleotide and amino acid metabolism, metabolism of other amino acids, glycan biosynthesis and metabolism, metabolism of cofactors and vitamins, metabolism of terpenoids and polyketides, biosynthesis of other secondary metabolites, and xenobiotic biodegradation and metabolism; (2) genetic information processing (846 sequences) including transcription, translation, folding, sorting and degradation, replication, and repair; (3) environmental information-processing pathways (358 sequences) including signal transduction, membrane transport, and signaling molecules and interaction; (4) cellular processes (264 sequences) including transport and catabolism; and (5) organismal systems (91 sequences) including immune system, endocrine system, development, and environmental adaptation. The KEGG metabolism pathway analysis also showed 63 sequences in eight different metabolism pathways including the glycolysis/gluconeogenesis pathway (n = 14), citrate cycle (TCA cycle) pathway (n = 11), pentose phosphate pathway (n = 6), pentose and glucuronate interconversions pathway (n = 9), fructose and mannose metabolism pathway (n = 9), galactose metabolism pathway (n = 6), ascorbate and aldarate metabolism pathway (n = 7), and fatty acid biosynthesis pathway (n = 1) (Table S2).
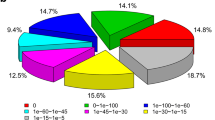
The 30,816 sequences were analyzed with the GO database (Fig. 2). In biological process, 5,656 sequences were expressed and the most expressed sequences are cellular process (3,364 sequences), developmental process (3,061 sequences), and multicellular organismal process (3,191 sequences). In cellular component, 4,719 sequences were expressed including cell part (4,459 sequences) and cell (4,466 sequences). In molecular function, 7,218 sequences were expressed including binding (4,597 sequences) and catalytic activity (3,084 sequences).

Functional annotations of the sequences from the fifth-stage larvae of A. cantonensis based on gene ontology categories. The pie charts show the general categories of biological process (a), cellular component (b), and molecular function (c)
In this NGS database, 4,719 sequences were expressed in cellular component including cytoskeletal-related genes. These highly expressed cytoskeletal-related genes are the tubulin-related genes (n = 32) including the tubulin alpha family (n = 11), tubulin gamma family (n = 3), tubulin folding cofactor E-Like protein (n = 4), and tubulin tyrosine ligase-like family (n = 14).
Molecules well represented in A. cantonensis in stress included antioxidants (n = 32), heat shock proteins (n = 112), and proteases (n = 153) (Fig. 3). The antioxidants highly expressed in A. cantonensis life cycle included the peroxiredoxin family (n = 5), thioredoxin family (n = 7), glutaredoxin family (n = 1), and glutathione transferases family (n = 19) (Table S3).

Distribution of stress-related proteins in the fifth-stage larvae of A. cantonensis
In addition to molecules related to stress, the NGS database also contained heat shock proteins (n = 112) including the heat shock factor (n = 5), heat shock protein 1 family (n = 11), heat shock protein 3 family (n = 7), heat shock protein 4 (n = 1), heat shock protein 6 family (n = 3), heat shock protein 12 family (n = 4), heat shock protein 16 family (n = 7), heat shock protein 17 family (n = 2), heat shock protein 25 family (n = 2), heat shock protein 43 family (n = 4), heat shock protein 60 family (n = 2), heat shock protein 70 family (n = 1), and DNaJ domain (prokaryotic heat shock protein) family (n = 63) (Table S4).
In this study, 153 proteases were found including the ADAM (disintegrin plus metalloprotease) family (n = 7), aspartyl protease family (n = 15), CED-3 protease suppressor family (n = 1), CLP protease family (n = 1), cysteine protease family (n = 10), inner mitochondrial membrane protease family (n = 6), intramembrane protease family (n = 7), nematode astacin protease family (n = 76), OTUBain deubiquitylating protease homolog family (n = 1), paraplegin AAA protease family (n = 3), trypsin-like protease family (n = 14), ubiquitin-like protease family (n = 10), YME1-like (yeast mitochondrial escape) AAA protease family (n = 1), and zinc metalloprotease family (n = 1) (Table S4).
Discussion
A. cantonensis is the rat lungworm. Human is a nonpermissive host, and the infection leads to eosinophilic meningitis or eosinophilic meningoencephalitis. After infecting the central nervous system of mice, A. cantonensis L5 induce a wide range of immune responses including eosinophilia recruitment and cytokine release (IL-4, IL-5, and eotaxin) via the NF-κB pathway (Lan et al. 2004). In addition, the migrating L5 have also been reported to cause mechanical injuries (Li et al. 2012). Blood–brain barrier dysfunction and CSF eosinophilia have also been observed in experimentally infected mice (Tsai et al. 2009). Therefore, studying A. cantonensis L5 gene expression in host is very important to understand the pathogenic mechanism.
Recent studies in high-throughput sequencing and bioinformatics provide researchers with the much needed tools to study nematodes (Jex et al. 2010; Webb and Rosenthal 2011), trematodes (Young et al. 2011), or other species (Vogel et al. 2011). NGS has been used to analyze Fasciola gigantica transcriptomes. From 20 million raw sequence reads, 30,000 contiguous sequences were assembled. These sequences were characterized on the amino acid level based on homology, gene ontology, and/or pathway mapping by KEGG (Young et al. 2011).
In our laboratory, 708 ESTs (159 clusters and 549 singletons) have been generated from A. cantonensis L5. In this study, we used NGS to study gene expression in the same developmental stage of this parasite. A total of 31,487 sequences were found and 14,509 (46.08 %) matched the C. elegans genome database. Therefore, we can perform a global study on the gene expression of A. cantonensis L5 in this study.
Proteins inferred from the transcriptome of A. cantonensis were compared with those predicted from transcriptomic data of 18 nematode ESTs from the NCBI database. Caenorhabditis species were found to be the most similar to A. cantonensis (C. elegans 97.87 %, C. brenneri 59.12 %, C. remanei 56.95 %). This result is very interesting since Caenorhabditis is a genus of nematodes living in bacteria-rich environments. In addition, it is free-living. C. elegans and Caenorhabditis briggsae are the most studied species in this genus. Moreover, C. elegans is an animal model and it is the first multicellular organism to have its genome completely sequenced and published in 1998. Gene expressions of blood-living parasite (B. malayi and O. volvulus) are also similar to A. cantonensis (52.35 %), since these parasites lived in blood vessels.
In the life cycle of A. cantonensis, the conversion of L3 to L5 is a complex differentiation process through regulating various proteins expression. During infection, A. cantonensis has to adapt to their new environment in a twofold way: L3 switch from a poikilothermic to a homoiothermic host and from a snail to a mammal. In addition, they are confronted with different types of host defense mechanisms both in snails and in mammals.
Albendazole is a member of the benzimidazole compounds and anticytoskeletal drugs. It has been used to treat a variety of worm infections (Barrère et al. 2012). This drug causes parasite tegument and intestine degenerative alteration by binding to the colchicine-sensitive site of tubulin, inhibiting tubulin polymerization and assembly into microtubules. In A. cantonensis, this drug has been reported to be the drug of choice against larvae and it is effective in the treatment of A. cantonensis infection in rabbits within 15 days after infection (Wang et al. 2006). Cell motility, growth, motor, and morphological changes in parasites require remodeling of cytoskeleton in response to intracellular and extracellular signals (Huang et al. 2009; O’Hagan et al. 2011; Martínez-Valladares et al. 2012). The reorganization of the microtubule network is important for the survival of nematodes (Banora et al. 2011). The identification of these cytoskeleton genes provided a rationale for the treatment of A. cantonensis.
Heat shock proteins are a highly conserved group of proteins in prokaryotic and eukaryotic organisms. They function as chaperones and assist newly synthesized protein folding (Morassutti et al. 2012). Their expressions are induced by the host immune responses such as reactive oxygen species (ROS) generation. After infection, ROS generation is induced in the mouse brain. It has been demonstrated that the expression of ROS-related enzymes such as glutathione reductase, glutathione peroxidase, and glutathione S-transferase (GST) significantly increases in the CSF of mice infected with A. cantonensis (Chung et al. 2010).
Antioxidants have been suggested to play a role in host immune modulation. They are highly expressed throughout the life cycle of A. cantonensis. Peroxiredoxin, thioredoxin, and glutathione transferases are important ones. Their expressions protect parasites from the attack of ROS generated by the host immune response (Dzik 2006; Sayed et al. 2006; Donnelly et al. 2008; Morassutti and Graeff-Teixeira 2012). We identified a total of 20 GSTs in the A. cantonensis L5 NGS database. GSTs are a large and important family of proteins found in many parasites to play a central role in H2O2 detoxification. Parasites contain multiple GSTs belonging to different GST classes and with differing enzyme activities to accommodate a wide range of functions of this enzyme family. GST in Fasciola hepatica influences host immune cell activities. It is present near the surface of the fluke and is expressed in eggs and newly excysted larvae as well as in the excretory/secretory fraction of the adults (LaCourse et al. 2012). In Opisthorchis viverrini, GST is one of the excretory/secretory products (ESP) and acts as a mitogen. It may also cause tumor development in the host. This antioxidant induces cell proliferation by activating pAKT and pERK (Daorueang et al. 2012).
In ESP of A. cantonensis, peroxiredoxin (Prx) is one of the highly expressed proteins (Donnelly et al. 2008). It is an enzyme reported to exist in many parasites and known to play a central role in H2O2 detoxification. In F. hepatica, Prx in the ESP has been shown to downregulate immune responses and to affect macrophage activation following injection into mice (Sayed et al. 2006). In our unpublished proteomic studies, highly expressed disulfide isomerase 1 and oxidoreductase have been determined by two-dimensional electrophoresis and mass spectrometry species-specific analysis and associated with defense oxidation stress. Inhibition of Prx expression in A. cantonensis not only causes the reduction of Th-2 immune responses of the host against F. hepatica and S. mansoni infection (Sayed et al. 2006) but also reduces their survival in the host (Donnelly et al. 2008).
Proteases are important for the survival of parasitic helminths by facilitating tissue penetration, digestion of food proteins, and immunomodulation. Moreover, the nutrition of blood-feeding nematodes depends on a multienzyme synergistic proteolytic cascade in the intestine, cleaving the ingested host hemoglobin tetramer into successively small fragments (Williamson et al. 2004). Aspartyl protease has been reported to initialize the degradation of host hemoglobin in blood-feeding parasite. It plays an important role in the nutrition of schistosomes (Brindley et al. 2001) and hookworms (Williamson et al. 2004). In our previous study, we identified an EST representing an aspartic protease from an A. cantonensis L5 dataset and this protease sequence encodes a protein with a predicted molecular mass of 46 kDa. Variations in the expression level of this aspartic protease gene exist at different development stages in both genders (Hwang et al. 2010). In other parasitic nematodes, highly expressed aspartyl protease has also been reported in Steinernema carpocapsae (Balasubramanian et al. 2012) and T. spiralis (Park et al. 2012).
By analysis of the ESTs of A. cantonensis L5, three cysteine protease genes have been reported. They are cathepsin B-like enzyme gene, 1, 2 (AC-cathB-1, AC-cathB-2), and hemoglobin-type cysteine protease gene (AC-hem) (Fang et al. 2010). A cysteine protease gene has also been isolated and described from the adult worm (Ni et al. 2012). Cysteine protease is a potential new target for the development of novel antimalarial drugs. It has reported that inhibitors of cysteine protease are able to block Plasmodium falciparum development in vitro and P. falciparum-infected mice could be cured (Sundararaj et al. 2012). In hookworm, a cysteine protease inhibitor may inhibit cysteine protease and has been used for the treatment of hookworm infection. This inhibitor may be developed as a novel anthelmintic to target or treat hookworm infection (Vermeire et al. 2012). RNA interference assays have been used to demonstrate functions of cysteine protease in B. malayi, and inhibition of cysteine protease causes the reduction in the numbers of secreted microfilariae in vitro. Moreover, siRNA-treated worms revealed a disruption in the process of embryogenesis (Ford et al. 2009).
This study is the first NGS-based study to set up a transcriptomic database of A. cantonensis L5 with identification and characterization of the genes. The results provide new insights into the survival, development, and host–parasite interactions of this blood-feeding nematode. Understanding of these aspects also allows us to carry out new investigations for the pathogenesis, clinical diagnosis, and chemotherapy of this zoonotic disease.
References
Aarthi S, Raj GD, Raman M, Blake D, Subramaniam C, Tomley F (2011) Expressed sequence tags from Eimeria brunette preliminary analysis and functional annotation. Parasitol Res 108:1059–1062
Alicata JE (1965) Biology and distribution of the rat lungworm, Angiostrongylus cantonensis, and its relationship to eosinophilic meningoencephalitis and other neurological disorders of man and animals. Adv Parasitol 3:223–248
Alicata JE (1988) Angiostrongyliasis cantonensis (eosinophilic meningitis): historical events in its recognition as a new parasitic disease of man. J Wash Acad Sci 78:38–46
Balasubramanian N, Toubarro D, Nascimento G, Ferreira R, Simões N (2012) Purification, molecular characterization and gene expression analysis of an aspartic protease (Sc-ASP113) from the nematode Steinernema carpocapsae during the parasitic stage. Mol Biochem Parasitol 182:37–44
Banora MY, Rodiuc N, Baldacci-Cresp F, Smertenko A, Bleve-Zacheo T, Mellilo MT, Karimi M, Hilson P, Evrard JL, Favery B, Engler G, Abad P, de Almeida Engler J (2011) Feeding cells induced by phytoparasitic nematodes require γ-tubulin ring complex for microtubule reorganization. PLoS Pathog 7:e1002343
Barrère V, Alvarez L, Suarez G, Ceballos L, Moreno L, Lanusse C (2012) Relationship between increased albendazole systemic exposure and changes in single nucleotide polymorphisms on the β-tubulin isotype 1 encoding gene in Haemonchus contortus. Vet Parasitol 186:344–349
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG et al (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53–59
Brindley PJ, Kalinna BH, Wong JY, Bogitsh BJ, King LT, Smyth DJ, Verity CK, Abbenante G, Brinkworth RI, Fairlie DP, Smythe ML, Milburn PJ, Bielefeldt-Ohmann H, Zheng Y, McManus DP (2001) Proteolysis of human hemoglobin by schistosome cathepsin D. Mol Biochem Parasitol 112:103–112
Campbell BE, Nagaraj SH, Hu M, Zhong W, Sternberg PW, Ong EK, Loukas A, Ranganathan S, Beveridge I, McInnes RL, Hutchinson GW, Gasser RB (2008) Gender-enriched transcripts in Haemonchus contortus—predicted functions and genetic interactions based on comparative analyses with Caenorhabditis elegans. Int J Parasitol 38:65–83
Cantacessi C, Campbell BE, Young ND, Jex AR, Hall RS, Presidente PJ, Zawadzki JL, Zhong W, Aleman-Meza B, Loukas A, Sternberg PW, Gasser RB (2010) Differences in transcription between free-living and CO2-activated third-stage larvae of Haemonchus contortus. BMC Genomics 11:266
Cantacessi C, Zou FC, Hall RS, Zhong W, Jex AR, Campbell BE, Ranganathan S, Sternberg PW, Zhu XQ, Gasser RB (2009) Bioinformatic analysis of abundant, gender-enriched transcripts of adult Ascaris suum (Nematoda) using a semi-automated workflow platform. Mol Cell Probes 23:205–217
Chang SH, Tang P, Wang LC (2011) A transcriptomic study on the pepsin-activated infective larvae of Angiostrongylus cantonensis. Mol Biochem Parasitol 179:47–50
Chen HT (1933) A preliminary report on a survey of animal parasites of Canton, China. Lingnan Sci J 12:65–74
Chen TW, Gan RC, Wu TH, Huang PJ, Lee CY, Chen YY, Chen CC, Tang P (2012) FastAnnotator—an efficient transcript annotation web tool. BMC Genomics 13(Suppl 7):S9
Cho PY, Lee MJ, Kim TI, Kang SY, Hong SJ (2006) Expressed sequence tag analysis of adult Clonorchis sinensis, the Chinese liver fluke. Parasitol Res 99:602–608
Cho PY, Kim TI, Whang SM, Hong SJ (2008) Gene expression profile of Clonorchis sinensis metacercariae. Parasitol Res 102:277–282
Chung LY, Chen CH, Wang LC, Chang SJ, Yen CM (2010) Oxidative stress in mice infected with Angiostrongylus cantonensis coincides with enhanced glutathione-dependent enzymes activity. Exp Parasitol 126:178–183
Daorueang D, Thuwajit P, Roitrakul S, Laha T, Kaewkes S, Endo Y, Thuwajit C (2012) Secreted Opisthorchis viverrini glutathione S-transferase regulates cell proliferation through AKT and ERK pathways in cholangiocarcinoma. Parasitol Int 61:155–161
Donnelly S, Stack CM, O’Neill SM, Sayed AA, Williams DL, Dalton JP (2008) Helminth 2-Cys peroxiredoxin drives Th2 responses through a mechanism involving alternatively activated macrophages. FASEB J 22:4022–4032
Droege M, Hill B (2008) The Genome Sequencer FLX System—longer reads, more applications, straight forward bioinformatics and more complete data sets. J Biotechnol 136:3–10
Dzik JM (2006) Molecules released by helminth parasites involved in host colonization. Acta Biochim Pol 53:33–64
Fang W, Xu S, Wang Y, Ni F, Zhang S, Liu J, Chen X, Luo D (2010) ES proteins analysis of Angiostrongylus cantonensis: products of the potential parasitism genes? Parasitol Res 106:1027–1032
Farias LP, Tararam CA, Miyasato PA, Nishiyama MY Jr, Oliveira KC, Kawano T, Verjovski-Almeida S, Leite LC (2011) Screening the Schistosoma mansoni transcriptome for genes differentially expressed in the schistosomulum stage in search for vaccine candidates. Parasitol Res 108:123–135
Ford L, Zhang J, Liu J, Hashmi S, Fuhrman JA, Oksov Y, Lustigman S (2009) Functional analysis of the cathepsin-like cysteine protease genes in adult Brugia malayi using RNA interference. PLoS Negl Trop Dis 3:e377
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I et al (2008) Single molecule DNA sequencing of a viral genome. Science 320:106–109
Huang CQ, Gasser RB, Cantacessi C, Nisbet AJ, Zhong W, Sternberg PW, Loukas A, Mulvenna J, Lin RQ, Chen N, Zhu XQ (2008) Genomic-bioinformatic analysis of transcripts enriched in the third-stage larva of the parasitic nematode Ascaris suum. PLoS Negl Trop Dis 2:e246
Huang KY, Chien KY, Lin YC, Hsu WM, Fong IK, Huang PJ, Yueh YM, Gan RR, Tang P (2009) A proteome reference map of Trichomonas vaginalis. Parasitol Res 104:927–933
Hwang KP, Chang SH, Wang LC (2010) Alterations in the expression level of a putative aspartic protease in the development of Angiostrongylus cantonensis. Acta Trop 113:289–294
Jacob J, Mitreva M, Vanholme B, Gheysen G (2008) Exploring the transcriptome of the burrowing nematode Radopholus similis. Mol Genet Genomics 280:1–17
Jex AR, Littlewood DT, Gasser RB (2010) Toward next-generation sequencing of mitochondrial genomes—focus on parasitic worms of animals and biotechnological implications. Biotechnol Adv 28:151–159
LaCourse EJ, Perally S, Morphew RM, Moxon JV, Prescott M, Dowling DJ, O’Neill SM, Kipar A, Hetzel U, Hoey E, Zafra R, Buffoni L, Pérez Arévalo J, Brophy PM (2012) The Sigma class glutathione transferase from the liver fluke Fasciola hepatica. PLoS Negl Trop Dis 6:e1666
Lan KP, Wang CJ, Hsu JD, Chen KM, Lai SC, Lee HH (2004) Induced eosinophilia and proliferation in Angiostrongylus cantonensis-infected mouse brain are associated with the induction of JAK/STAT1, IAP/NF-kappaB and MEKK1/JNK signals. J Helminthol 78:311–317
Lee JS, Lee J, Park SJ, Yong TS (2003) Analysis of the genes expressed in Clonorchis sinensis adults using the expressed sequence tag approach. Parasitol Res 91:283–289
Li Q, Zhao Y, Ni B, Yao C, Zhou Y, Xu W, Wang Z, Qiao Z (2008) Comparison of the expression profiles of promastigotes and axenic amastigotes in Leishmania donovani using serial analysis of gene expression. Parasitol Res 103:821–828
Li ZY, Sun R, Li J, Song YX, Lin YC, Zeng X (2012) Efficacy of albendazole combined with a marine fungal extract (m2-9) against Angiostrongylus cantonensis-induced meningitis in mice. J Helminthol 86:410–417
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Martínez-Valladares M, Donnan A, Geldhof P, Jackson F, Rojo-Vázquez FA, Skuce P (2012) Pyrosequencing analysis of the beta-tubulin gene in Spanish Teladorsagia circumcincta field isolates. Vet Parasitol 184:371–376
Morassutti AL, Graeff-Teixeira C (2012) Interface molecules of Angiostrongylus cantonensis: their role in parasite survival and modulation of host defenses. Int J Inflamm 2012:512097
Morassutti AL, Levert K, Pinto PM, da Silva AJ, Wilkins P, Graeff-Teixeira C (2012) Characterization of Angiostrongylus cantonensis excretory-secretory proteins as potential diagnostic targets. Exp Parasitol 130:26–31
Nagaraj SH, Deshpande N, Gasser RB, Ranganathan S (2007a) ESTExplorer: an expressed sequence tag (EST) assembly and annotation platform. Nucleic Acids Res 35:143–147
Nagaraj SH, Gasser RB, Ranganathan S (2007b) A hitchhiker’s guide to expressed sequence tag (EST) analysis. Brief Bioinform 8:6–21
Ni F, Wang Y, Zhang J, Yu L, Fang W, Luo D (2012) Cathepsin B-like and hemoglobin-type cysteine proteases: stage-specific gene expression in Angiostrongylus cantonensis. Exp Parasitol 131:433–441
Nisbet AJ, Cottee PA, Gasser RB (2008) Genomics of reproduction in nematodes: prospects for parasite intervention? Trends Parasitol 24:89–95
O’Hagan R, Piasecki BP, Silva M, Phirke P, Nguyen KC, Hall DH, Swoboda P, Barr MM (2011) The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr Biol 21:1685–1694
Pandey V, Nutter RC, Prediger E (2008) Applied Biosystems SOLiD™ System: ligation-based sequencing. In: Jantz M (ed) Next generation genome sequencing: towards personalized medicine. Wiley, Weinheim, pp 29–42
Park JN, Park SK, Cho MK, Park MK, Kang SA, Kim DH, Yu HS (2012) Molecular characterization of 45 kDa aspartic protease of Trichinella spiralis. Vet Parasitol 190:510–518
Peng HJ, Chen XG, Wang XZ, Lun ZR (2003) Analysis of the gene expression profile of Schistosoma japonicum cercariae by a strategy based on expressed sequence tags. Parasitol Res 90:287–293
Punyagupta S, Juttijudata P, Bunnag T (1975) Eosinophilic meningitis in Thailand. Clinical studies of 484 typical cases probably caused by Angiostrongylus cantonensis. Am J Trop Med Hyg 24:921–931
Ranganathan S, Menon R, Gasser RB (2009) Advanced in silico analysis of expressed sequence tag (EST) data for parasitic nematodes of major socioeconomic importance—fundamental insights toward biotechnological outcomes. Biotechnol Adv 27:439–448
Sayed AA, Cook SK, Williams DL (2006) Redox balance mechanisms in Schistosoma mansoni rely on peroxiredoxins and albumin and implicate peroxiredoxins as novel drug targets. J Biol Chem 281:17001–17010
Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26:1135–1145
Sundararaj S, Singh D, Saxena AK, Vashisht K, Sijwali PS, Dixit R, Pandey KC (2012) The ionic and hydrophobic interactions are required for the auto activation of cysteine proteases of Plasmodium falciparum. PLoS One 7:e47227
Tsai HC, Huang YL, Liu YC, Wann SR, Lee SS, Chen ER (2009) Dynamic changes of hepatocyte growth factor in eosinophilic meningitis caused by Angiostrongylus cantonensis infection. Am J Trop Med Hyg 80:980–982
Vermeire JJ, Lantz LD, Caffrey CR (2012) Cure of hookworm infection with a cysteine protease inhibitor. PLoS Negl Trop Dis 6:e1680
Vogel H, Altincicek B, Glöckner G, Vilcinskas A (2011) A comprehensive transcriptome and immune-gene repertoire of the lepidopteran model host Galleria mellonella. BMC Genomics 12:308
Wang LC, Chao D, Chen ER (1989) Acquired immunity in rats against Angiostrongylus cantonensis infection. Int J Parasitol 19:617–620
Wang LC, Jung SM, Chen CC, Wong HF, Wan DP, Wan YL (2006) Pathological changes in the brains of rabbits experimentally infected with Angiostrongylus cantonensis after albendazole treatment: histopathological and magnetic resonance imaging studies. J Antimicrob Chemother 57:294–300
Wang QP, Lai DH, Zhu XQ, Chen XG, Lun ZR (2008) Human angiostrongyliasis. Lancet Infect Dis 8:621–630
Wang QP, Wu ZD, Wei J, Owen RL, Lun ZR (2012) Human Angiostrongylus cantonensis: an update. Eur J Clin Microbiol Infect Dis 31:389–395
Webb KM, Rosenthal BM (2011) Next-generation sequencing of the Trichinella murrelli mitochondrial genome allows comprehensive comparison of its divergence from the principal agent of human trichinellosis, Trichinella spiralis. Infect Genet Evol 11:116–123
Williamson AL, Lecchi P, Turk BE, Choe Y, Hotez PJ, McKerrow JH, Cantley LC, Sajid M, Craik CS, Loukas A (2004) A multi-enzyme cascade of hemoglobin proteolysis in the intestine of blood-feeding hookworms. J Biol Chem 279:35950–35957
Xu SS, Ni F, Luo DM (2009) Expressed sequence tags (ESTs) analysis of Angiostrongylus cantonensis. Chin J Parasitol Parasit Dis 27:248–250
Young ND, Jex AR, Cantacessi C, Hall RS, Campbell BE, Spithill TW (2011) A portrait of the transcriptome of the neglected trematode, Fasciola gigantica—biological and biotechnological implications. PLoS Negl Trop Dis 5:e1004
Acknowledgments
This work was supported in part by grants from the National Science Council, Executive Yuan, ROC (NSC100-2320-B-182-013 and NSC101-2320-B-182-045) and Chang Gung Memorial Hospital Research Grant CMRPD1B0351. We thank the Chang Gung Molecular Medicine Research Center for the technical supports.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Lian-Chen Wang and Kuang-Yao Chen contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Similarities of transcripts of Angiostrongylus cantonensis fifth-stage larvae to Caenorhabditis elegans based on BLASTx. (XLS 7156 kb)
Table S2
Metabolic pathways in the fifth-stage larvae of Angiostrongylus cantonensis mapped by Kyoto Encyclopedia of Genes and Genomes (KEGG). (XLS 61 kb)
Table S3
ROS related genes in the fifth-larva of Angiostrongylus cantonensis NGS database. (XLS 40 kb)
Table S4
Heat shock protein related genes in fifth larva of Angiostrongylus cantonensis NGS database. (XLS 57 kb)
Table S5
Protease related genes in fifth larva of Angiostrongylus cantonensis NGS database. (XLS 68 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Wang, LC., Chen, KY., Chang, SH. et al. Transcriptome profiling of the fifth-stage larvae of Angiostrongylus cantonensis by next-generation sequencing. Parasitol Res 112, 3193–3202 (2013). https://doi.org/10.1007/s00436-013-3495-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-013-3495-z