
Abstract
Purpose
A wide therapeutic repertoire has become available to oncologists including radio- and chemotherapy, small molecules and monoclonal antibodies. However, drug efficacy can be limited by genetic heterogeneity. Here, we designed a webtool that facilitates the data analysis of the in vitro drug sensitivity data on 265 approved compounds from the GDSC database in association with a plethora of genetic changes documented for 1001 cell lines in the CCLE data.
Methods
The webtool computes odds ratios of drug resistance for a queried set of genetic alterations. It provides results on the efficacy of single compounds or groups of compounds assigned to cellular signaling pathways. Webtool availability: https://tools.hornlab.org/GDSC/.
Results
We first replicated established associations of genetic driver mutations in BRAF, RAS genes and EGFR with drug response. We then tested the ‘BRCAness’ hypothesis and did not find increased sensitivity to the assayed PARP inhibitors. Analyzing specific PIK3CA mutations related to cancer and mendelian overgrowth, we found support for the described sensitivity of H1047 mutants to GSK690693 targeting the AKT pathway. Testing a co-mutated gene pair, GATA3 activation abolished PTEN-related sensitivity to PI3K/mTOR inhibition. Finally, the pharmacogenomic modifier ABCB1 was associated with olaparib resistance.
Conclusions
This tool could identify potential drug candidates in the presence of custom sets of genetic changes and moreover, improve the understanding of signaling pathways. The underlying computer code can be adapted to larger drug response datasets to help structure and accommodate the increasingly large biomedical knowledge base.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
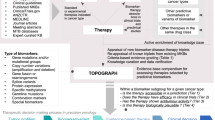
Genetic diversity across cancers modifies the tumor’s drug responsiveness and impedes the use of widely applicable drugs without patient stratification (Lin and Sheltzer 2020). The BRAF V600-specific inhibition of tumor growth across various tumor entities is a prime example of personalized cancer treatment (Subbiah et al. 2020) and depends on the availability of diagnostic biomarkers with predictive power for drug response (Chin et al. 2011). To identify and establish those powerful decisive markers, it is crucial to associate specific genetic changes with drug response data. Complex datasets have been generated for cancer cell lines to provide information on genetic changes and drug response. The Genomics of Drug Sensitivity in Cancer Project (GDSC) and The Cancer Cell Line Encyclopedia (CCLE) represent two of such databases (Iorio et al. 2016; Ghandi et al. 2019). While GDSC provides drug response data including bioinformatically estimated concentration–response curves, IC50 values as well as tested genomic associations, the CCLE dataset holds information on altered gene copy number, point mutations, mRNA levels, and gene fusions. Although public webtools were developed for ready access and analysis of the above datasets (Cerami et al. 2012; Yang et al. 2013; Piñeiro-Yáñez et al. 2018; Basu et al. 2013; Najgebauer et al. 2020), these tools often lack the possibility to analyze a custom set of mutations or a combination of co-occurring gene alterations. Here, we designed a straightforward publicly accessible tool that combines the GDSC drug response data and genetic data collected for the CCLE samples. It enables researchers to query individual combinations of genetic alterations to assess drug sensitivity for a larger variety of cell lines (Fig. 1A). Our approach allows any combination of genetic changes in the query genes to address cellular pathways. Since different genetic changes of the same gene can mediate various functional effects, we offer custom queries for hypothesis testing and discovery of drugs for potential repurposing.

Workflow of the webtool and verification of the BRAF–MAPK pathway association. A Users define custom genetic changes and test across cell lines from various cancer entities. Categorization in ‘resistant’ or ‘sensitive’ from Iorio et al. (Iorio et al. 2016): cell viability measured using the CellTiter-Glo® Assay, IC50 values subsequently estimated by a non-linear mixed effects model, binarization threshold for each drug. B Principle of testing the gain of function (GOF) BRAF V600-mutated cell lines revealing moderate resistance to EGFR and strong sensitivity to ERK/MAPK pathway compounds. Odds ratios (OR) for resistance in the altered cell lines (mut) above each bar plot (Fisher’s tests). Numbers of cell lines pooled in each pathway category differ by available data indicated in the bars. Using Bonferroni correction for testing 22 pathways, p-values < 0.0023 are considered significant
Materials and methods
Implementation
The webtool was implemented using R shiny in R version 4.0.5. To avoid duplications of 15 compounds screened twice, the screenings with higher data availability were used. Drugs were assigned to one of 22 target pathways as previously described (Iorio et al. 2016). Further information on compounds can be retrieved in the GDSC database at https://www.cancerrxgene.org/ and the compounds of the GDSC1 dataset are included in our webtool. Here, we analyzed genes across all available cell lines as the majority of cell lines were profiled for the queried alterations. The ‘BRCAness’ analysis was additionally performed on breast cancer cell lines only. Users can specify which genetic changes and cell lines shall be included in the analysis, for every gene the type of genetic changes can be selected independently. Furthermore, cell lines with missing data, which were not profiled for a specific type of alteration, can be excluded to avoid ascertainment biases in the analyses, e.g., of gene fusions which have not been profiled for the majority of cell lines (Fig. 2). Plots and data tables are provided for download.

Step-by-step user workflow. A Obtain genetic alterations of query genes in the ‘alterations_across_samples.tsv’ file from www.cBioPortal.org as described at https://tools.hornlab.org/GDSC/Manual.pdf and in Supplementary Fig. 1–2. B Based on this file, cell lines are classified in ‘genetically changed’ and ‘wild type’ as reference. Option 1: tumor entities (tissue origins) of interest can be selected. Up to three genes can be chosen for analysis. The webtool provides automated assignment of cell lines into groups, by default ‘presumably activating’: amplifications (CNV up), high mRNA as defined by cBioportal. Default: ‘presumably deactivating’: homozygous deletions (CNV down), low mRNA, nonsense- and frameshift mutations. Users can additionally select missense mutations, splice variants and gene fusions. For two or three genes, cell lines can be defined as ‘mutated’ if at least one of the genes is changed or optionally, if all genes are changed (co-mutation). Option 2: custom grouping based on a tab-delimited text file and used in the upload option of the webtool. It should contain 3 columns named ‘Sample.ID’, ‘Patient.ID’, and ‘changed’. The first two columns are retrieved from the ‘alterations_across_sample.tsv’ file and the third column, added by the user, defines the cell lines as ‘mut’ for mutated/genetically changed and ‘wt’ for wild type (empty cells to exclude cell lines)
Statistical analysis
From the contingency tables of genetically changed and wild-type groups, odds ratios (OR) for resistance and Fisher’s exact tests (two-sided) were computed, within each pathway as well as for every single compound. Bonferroni-corrected p value thresholds are < 0.0023 testing 22 pathways and p < 0.0002 testing 250 drugs. For counts of zero, it was not possible to calculate OR and ratios of resistant and sensitive cell lines are shown in percent above the bar plot.
Results and discussion
As a proof of principle, we tested established gene–drug associations across tumor entities. Specific gene mutations are routinely used as biomarkers to guide targeted therapies, such as BRAF V600 mutations for the targeted MAP kinase inhibition (MAPKi) therapy of cancer patients (Subbiah et al. 2020). We replicated this established association with strong statistical support in our webtool. BRAF V600-positive cell lines were sensitive to compounds targeting the ERK–MAPK signaling pathway (OR 0.16, p = 3.2e−125, Fig. 1B) such as PLX4720 (OR 0.03, p = 6.2e−38) and dabrafenib (OR 0.02, p = 3.87e−36, Supplementary Fig. 3). Of note, BRAF V600-mutated cell lines were resistant to compounds targeting the PI3K/mTOR pathway (OR 1.72, p = 1.12e−7), EGFR pathway (OR 3.10, p = 2.37e−7), and the RTK pathway (OR 1.82, p = 1.47e−9, Supplementary Fig. 4A).
Since RAS proteins activate the MAPK-signaling pathway (Bonni et al. 1999), we expected cancer cell lines carrying one of the recurrent point mutations in NRAS, HRAS or KRAS to be sensitive to compounds targeting the ERK–MAPK signaling pathway as well. This alleged association was confirmed across entities with great statistical support (OR 0.49, p = 3.71e−33, Supplementary Fig. 4B). As the role of EGFR and ERBB2 overexpression and amplification in cancer is well described (Olayioye et al. 2000), we expected to replicate a sensitivity to compounds inhibiting the EGFR-signaling pathway in EGFR- or ERBB2-altered cell lines. Cell lines with potentially activating changes including gene amplifications as well as high mRNA levels in either EGFR or ERBB2, or both genes were associated with sensitivity to EGFR pathway compounds (OR 0.51, p = 3.05e−8, Supplementary Fig. 5). Compounds that showed sensitivity in EGFR/ERBB2-altered cell lines almost exclusively target the EGFR pathway (Fig. 3A). Furthermore, as PTEN loss results in hyperactive PI3K signaling (Carracedo and Pandolfi 2008) we tested cell lines with PTEN deactivating changes (homozygous deletions, splice variants, frameshift and nonsense mutations) and confirmed those to be sensitive to compounds inhibiting the PI3K/mTOR pathway (OR 0.75, p = 2.94e−5, Supplementary Fig. 6A, B). Hence, we replicated several established associations of genetic changes and drug sensitivity across cancer entities, indicating the reliability of the presented webtool to detect true strong associations.

Verification of EGFR pathway association and new drug dependencies. A Compounds that show activity in EGFR/ERBB2-activated cell lines almost exclusively belong to the EGFR pathway. Forest plot with OR > 1 shows resistance of altered cell lines to drugs, OR < 1 indicates sensitivity. Compounds shown if p < 0.05 from Fisher’s exact tests for candidate approaches. For discovery testing 250 compounds, Bonferroni-corrected p < 0.0002 are considered significant. B Within frequent PIK3CA point mutations only H1047 mutant cell lines were sensitive to AKT-inhibitor GSK690693 (Supplementary Table 3). Frequencies and image for CCLE 2019 from www.cbioportal.org. Colored regions: protein domains. Light green: missense variants of unknown significance, dark green: driver missense mutations. C GATA3 activation and abolished PTEN-related sensitivity to PI3K/mTOR inhibition. D Model of olaparib exclusion with activated ABCB1. GOF/LOF: gain/loss of function
Next, we tested proposed gene–drug associations, at first the ‘BRCAness’ hypothesis (Lord and Ashworth 2016), comparing cancer cell lines with deactivating changes (homozygous deletions, splice variants, frame shift, and nonsense mutations) in at least one of the following ‘BRCAness’ genes: BRCA1, BRCA2, ATM, ATR, RAD51C, and RAD51D with wild types across entities. The sensitizing effect of BRCA deactivation for poly (ADP-ribose) polymerase inhibition (PARPi) therapy has been shown multiple times across cancer entities (Fong et al. 2009). However, we found no evidence for an increased sensitivity to the assayed PARP inhibitors neither across entities nor in breast cancer cell lines (Supplementary Table 2). These results indicate that ‘BRCAness’ may not be an obligate prerequisite for benefit from PARPi in line with recent studies (Shen et al. 2019). As a second test of a previously reported association we tested PI3K/AKT/mTOR inhibitors and PIK3CA mutations. Somatic mutations have been reported to occur in cancer as well as pathogenic germline variants in PIK3CA-related overgrowth. We tested a set of 21 drugs from the group of PI3K/AKT/mTOR inhibitors and observed support for the described sensitivity of the H1047 mutant cell lines (Janku et al. 2013) for at least one inhibitor in the pathway, the AKT-inhibitor GSK690693 (OR 0.21, p = 0.00028 at alpha = 0.002, Fig. 3B, Supplementary Table 3). Furthermore, PIK3CA E545K or E542K mutated cancer cell lines had been described to be less sensitive (Janku et al. 2013) and indeed were not associated with an apparent response to GSK690693 (OR 0.81, p = 0.69). Beyond cancer, the mutational spectra of PIK3CA-related cancer and PIK3CA-related overgrowth syndromes (PROS) overlap (Venot et al. 2018). Patients with PROS showed improvements in disease symptoms following treatment with the PI3K inhibitor BYL719 and we, thus, tested the most frequent PROS PIK3CA mutations H1047, E542 and C420. Although BYL719 is not included in our drug dataset, cell lines harboring one of the 3 mutations also showed sensitivity to the Akt inhibitor GSK690693 (OR 0.28, p = 0.00019). Hence, we support the notion that blocking Akt is efficient in the presence of one of the three most frequent PIK3CA-overgrowth mutations.
To test drug resistance of co-mutations in a large cancer entity we chose the significantly co-mutated gene pair PTEN and GATA3 (co-mutation p < 0.001, Supplementary Fig. 6C–E). GATA3 expression was described to delay tumor progression and reduce Akt activation in PTEN-deficient mouse prostate cancer (Nguyen et al. 2013) and resulted in differential drug sensitivity in breast cancer (Mair et al. 2016). While here, PTEN deactivation was again associated with the previously observed sensitivity to compounds targeting the PI3K/MTOR pathway, a concomitant GATA3 activation (amplification, mRNA high) conferred resistance (OR 1.59, p = 0.0018, Fig. 3C, Supplementary Table 4). A concomitant GATA3 deactivation (homozygous deletions, splice variants, frameshift and nonsense mutations) did not yield a significant result (OR 1.38, p = 0.029, significance level testing 22 pathways: 0.002) although an opposite effect could be expected from GATA3 deactivation leading to either no changes in PI3K/mTOR inhibition retaining sensitivity or even enhancing sensitivity. These results may prompt more experiments to test if GATA3 is indeed functionally involved in the PTEN-axis of PI3K/mTOR inhibitor sensitivity.
Next, we tested putatively activating genetic changes of the pharmacogenomic modifier ABCB1 vs. the wild type and found resistance to compounds from various pathways (Supplementary Table 5). Olaparib efflux has been reported for advanced prostate cancer over-expressing ABCB1 (Lombard et al. 2019) and also here all of the ABCB1-activated cell lines were resistant to olaparib (n = 76, p = 0.00015, Fig. 3D), while 12.4% of wild-type cell lines were sensitive. A deactivation of ABCB1 in 21 cell lines had no significant association with olaparib efficacy (OR 0.82, p = 0.7373).
Conclusion
In conclusion, we present a powerful webtool to analyze associations of genetic alterations and drug response from the CCLE and GDSC datasets and enable fast hypothesis testing and discovery approaches across and within cancer entities. The swift analysis of molecular tumor heterogeneity may help to identify compounds for drug repurposing and prioritization and to understand settings of co-mutation. More broadly, beyond cancer it could be applied to investigate mechanisms of cell signaling pathways. As the public data sets are currently growing fast, larger sample sizes will improve statistical power, McInnes et al. recently released a pharmacogenetic analysis including 487,409 participants (McInnes, Lavertu et al. 2021). The open source of our webtool is technically applicable to these larger datasets with binary and continuous measures for drug response. Hence, it will be possible to further expand it as a functional tool to bridge from big drug sensitivity screens to bench-side researchers.
Data availability
Generated code, group definitions of cell lines and resulting tables are available at https://github.com/MyriamBoeschen/Drug_Response_Tool, data sources in Supplementary Table 1.
References
Basu A, Bodycombe NE, Cheah JH, Price EV, Liu Ke, Schaefer GI, Ebright RY et al (2013) An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154(5):1151–1161. https://doi.org/10.1016/j.cell.2013.08.003
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286(5443):1358–1362. https://doi.org/10.1126/science.286.5443.1358
Carracedo A, Pandolfi PP (2008) The PTEN–PI3K pathway: of feedbacks and cross-talks. Oncogene 27(41):5527–5541. https://doi.org/10.1038/onc.2008.247
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2(5):401–404. https://doi.org/10.1158/2159-8290.CD-12-0095
Chin L, Andersen JN, Andrew Futreal P (2011) Cancer genomics: from discovery science to personalized medicine. Nat Med 17(3):297–303. https://doi.org/10.1038/nm.2323
Fong PC, Boss DS, Yap TA, Tutt A, Peijun Wu, Mergui-Roelvink M, Mortimer P et al (2009) Inhibition of poly (ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361(2):123–134. https://doi.org/10.1056/NEJMoa0900212
Ghandi M, Franklin WH, Judit J-V, Gregory VK, Christopher CL, Mcdonald ER 3rd, Jordi B et al (2019) Next-generation characterization of the cancer cell line encyclopedia. Nature 569(7757):503–8. https://doi.org/10.1038/s41586-019-1186-3
Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N et al (2016) A landscape of pharmacogenomic interactions in cancer. Cell 166(3):740–754. https://doi.org/10.1016/j.cell.2016.06.017
Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, Siqing Fu et al (2013) PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Can Res 73(1):276–284. https://doi.org/10.1158/0008-5472.CAN-12-1726
Lin A, Sheltzer JM (2020) Discovering and validating cancer genetic dependencies: approaches and pitfalls. Nat Rev Genet 21(11):671–682. https://doi.org/10.1038/s41576-020-0247-7
Lombard AP, Liu C, Armstrong CM, D’Abronzo LS, Lou W, Chen H, Dall’Era M, Ghosh PM, Evans CP, Gao AC (2019) Overexpressed ABCB1 induces olaparib-taxane cross-resistance in advanced prostate cancer. Transl Oncol 12(7):871–78. https://doi.org/10.1016/j.tranon.2019.04.007
Lord CJ, Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16(2):110–120. https://doi.org/10.1038/nrc.2015.21
Mair B, Konopka T, Kerzendorfer C, Sleiman K, Salic S, Serra V, Muellner MK, Theodorou V, Nijman SMB (2016) Gain- and loss-of-function mutations in the breast cancer gene GATA3 result in differential drug sensitivity. PLoS Genet 12(9):e1006279. https://doi.org/10.1371/journal.pgen.1006279
McInnes G, Lavertu A, Sangkuhl K, Klein TE, Whirl-Carrillo M, Altman RB (2021) “Pharmacogenetics at Scale: An Analysis of the UK Biobank.” Clin Pharmacol Therapeut 109(6):1528–37. https://doi.org/10.1002/cpt.2122
Najgebauer H, Yang Mi, Francies HE, Pacini C, Stronach EA, Garnett MJ, Saez-Rodriguez J, Iorio F (2020) CELLector: genomics-guided selection of cancer in vitro models. Cell Syst 10(5):424–32.e6. https://doi.org/10.1016/j.cels.2020.04.007
Nguyen AHT, Tremblay M, Haigh K, Koumakpayi IH, Paquet M, Pandolfi PP, Mes-Masson A-M, Saad F, Haigh JJ, Bouchard M (2013) Gata3 antagonizes cancer progression in Pten-deficient prostates. Hum Mol Genet 22(12):2400–2410. https://doi.org/10.1093/hmg/ddt088
Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19(13):3159–3167. https://doi.org/10.1093/emboj/19.13.3159
Piñeiro-Yáñez E, Reboiro-Jato M, Gómez-López G, Perales-Patón J, Troulé K, Rodríguez JM, Tejero H et al (2018) PanDrugs: a novel method to prioritize anticancer drug treatments according to individual genomic data. Genome Med 10(1):41. https://doi.org/10.1186/s13073-018-0546-1
Shen J, Zhao W, Zhenlin Ju, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G (2019) PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Can Res 79(2):311–319. https://doi.org/10.1158/0008-5472.CAN-18-1003
Subbiah V, Puzanov I, Blay J-Y, Ian Chau A, Lockhart C, Raje NS, Wolf J et al (2020) Pan-cancer efficacy of vemurafenib in -mutant non-melanoma cancers. Cancer Discov 10(5):657–663. https://doi.org/10.1158/2159-8290.CD-19-1265
Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong J-P, Blanc E et al (2018) Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 558(7711):540–546. https://doi.org/10.1038/s41586-018-0217-9
Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N et al (2013) Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41(4):D955-61. https://doi.org/10.1093/nar/gks1111
Acknowledgements
We thank Udo Stenzel for advice on the rationale and implementation of this webtool.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) HO 6389/2-2, ‘KFO 337’—405344257 to SH and SFB 1052 Project B10 to DLD. The funders were not involved in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MB: conceptualization, data curation, software, formal analysis, validation, investigation, visualization, methodology, writing—original draft. DLD: conceptualization, writing—review and editing. MS: conceptualization, writing—review and editing. ML: conceptualization, writing—review and editing. TS: conceptualization, supervision, funding acquisition, writing—review and editing. SH: conceptualization, methodology, formal analysis, validation, investigation, visualization, supervision, project administration, funding acquisition, writing—review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boeschen, M., Le Duc, D., Stiller, M. et al. Interactive webtool for analyzing drug sensitivity and resistance associated with genetic signatures of cancer cell lines. J Cancer Res Clin Oncol 149, 5539–5545 (2023). https://doi.org/10.1007/s00432-022-04503-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-022-04503-2