
Abstract
Lipopolysaccharide (LPS)-responsive beige-like anchor (LRBA) protein deficiency is a rare syndrome of primary immune deficiency and immune dysregulation. In this study, we sought to summarize our experience with respiratory manifestations in LRBA-deficient patients. We conducted a retrospective analysis of the medical records of LRBA-deficient patients treated at Hadassah-Hebrew University Medical Center, Jerusalem, Israel. Data retrieved included pulmonary workup, disease course, treatment, and outcome. Ten patients were included. Mean age at presentation of LRBA deficiency-related symptoms was 4.65 years (range 3 months–14 years). Respiratory symptoms were noted in six patients and consisted of chronic cough. Computed tomography revealed consolidation in five patients, atelectasis and bronchiectasis in two patients each, and diffuse interstitial lung disease in two additional patients. Respiratory tract cultures yielded a bacterial pathogen in five patients. Seven patients required active therapy: intravenous immunoglobulins (six patients), immunosuppressive drugs (five patients), and one was successfully treated with abatacept. Two patients underwent successful bone marrow transplantation. Mean follow-up period was 4.5 (range 0.4–14.4) years. On their latest examination, seven patients had no respiratory symptoms.
Conclusion: Pulmonary manifestations are common in LRBA deficiency. Respiratory characteristics in LRBA-deficient patients should be investigated, monitored, and treated from the time of diagnosis.
What is Known: • Lipopolysaccharide-responsive beige-like anchor (LRBA) deficiency is a syndrome of primary immune deficiency and immune dysregulation. • Studies concerning the pulmonary characteristics of LRBA-deficient patients are lacking. | |
What is New: • Respiratory manifestations include infections, bronchiectasis, interstitial lung disease, thoracic lymphadenopathy, and clubbing. • Awareness to pulmonary morbidity in LRBA-deficient patients and involvement of a pulmonologist in the workup and clinical decision-making is important. • Respiratory characteristics in LRBA-deficient patients should be investigated, monitored, and treated from a young age. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lipopolysaccharide (LPS)-responsive beige-like anchor (LRBA) protein belongs to the BEACH-WD40 protein family [2]. Located on 4q31.3, the LRBA gene is expressed in a variety of organs, including the brain, heart, placenta, liver, skeletal muscles, kidneys, and pancreas [2, 19]. LRBA protein plays an important role in the expression of cytotoxic T-lymphocyte antigen (CTLA)-4 in CD4+ and FOXP3+ T cells [11, 14, 18].
LRBA deleterious mutations are associated with a syndrome of primary immune deficiency (PID) comprised of a combined immunodeficiency, autoimmune cytopenia, recurrent infections, immune dysregulation, hepatosplenomegaly, and enteropathy [4, 12, 13, 16, 18, 19].
Studies describing patients with LRBA deficiency focus on immunological and hematological characteristics [4, 12,13,14, 18, 19]. However, detailed descriptions of pulmonary manifestations in these patients are lacking.
In this study, we summarize our experience with respiratory manifestations of 10 LRBA-deficient patients. We present pulmonary diagnostic workup, clinical course and treatment, and review current literature.
Materials and methods
Patients
The study was conducted at Hadassah-Hebrew University Medical Center, Jerusalem, Israel. All patients that were diagnosed with LRBA deficiency, due to a mutation in the LRBA gene, and treated between January 2000 and February 2018 were included in the study. LRBA gene mutations were identified using whole exome sequencing (WES) and were confirmed by Sanger sequencing. The method of WES used in our study was previously described [22]. Furthermore, patients P1–P4 have been previously reported [13, 22].
Study design
This is a retrospective analysis of the medical records of LRBA-deficient patients. Data collected included clinical presentation, course, treatment, and outcome. Results of the pulmonary workup including fiber-optic bronchoscopy studies, bronchoalveolar lavage (BAL) cultures, pulmonary function testing (PFT), chest X-rays (CXR), and computed tomography (CT) scans were reviewed and analyzed. One patient (P6) was summoned to a follow-up visit and completed PFT. CXR and CT scans were reviewed by a senior pediatric radiologist. PFT and bronchoscopy studies were reviewed by a senior pediatric pulmonologist.
Pulmonary function testing, computed tomography scans, and bronchoscopy studies
PFT was performed according to the American Thoracic Society/European Respiratory Society guidelines [20]. Evaluations included spirometry, lung volumes by plethysmography, airway resistance, and diffusion capacity corrected for hemoglobin and lung volumes. Values are presented as percent predicted values. PFT reference values have considered failure to thrive (FTT) and growth retardation.
CT scans were performed, using Phillips 256-slice scanner (Brilliance iCT, Philips), utilizing standard protocol appropriate to the patient weight, with contrast injection.
Bronchoscopy was performed using a pediatric flexible bronchoscope under general anesthesia. BAL aliquots of ~ 1 ml/kg normal saline were used in the right middle lobe and lingula in addition to areas of evident airway secretions. All samples were sent for Gram and acid-fast stains, fungal smear, bacterial, fungal, and mycobacterial cultures. One patient (P7) underwent a trans-bronchial biopsy with multiple specimens obtained under fluoroscopy.
Ethical review of the study
The study was approved by the Hadassah and Israeli Ministry of Health institutional review boards (number 187-17-HMO).
Results
Clinical characteristics of the patients
Clinical characteristics are presented in Table 1. Ten Arab patients (eight males), representing six consanguineous families, were included in our study. P1, P2, and P3 are siblings of one family; P7, P8, and P9 are siblings of a separate family. Mean patient age (excluding P3 and P9) at first clinical presentation related to LRBA deficiency was 4.65 (range 0.25–14) years.
Autoimmune manifestations on initial presentation included ulcerative colitis (P10), immune thrombocytopenic purpura (P1, P2, P4, P7, and P10), autoimmune hemolytic anemia (P1, P2, P4, and P6), and Graves’ disease (P8). FTT was noted in one patient (P7). A history of recurrent infections was noted in four patients: Three patients had recurrent pneumonia (P1, P5, and P7), one patient (P6) had recurrent urinary tract infections, and another patient (P7) had recurrent acute otitis media. Two patients suffered from chronic diarrhea (P5 and P6). Physical examination revealed lymphadenopathy and splenomegaly in seven (P1, P2, P4, P5, P6, P7, and P10) and five patients (P2, P4, P5, P6, and P7), respectively.
Initial laboratory workup
Complete blood count on their first admission demonstrated thrombocytopenia in four patients (P1, P2, P4, and P10; 10,000, 13,000, 71,000, and 8000 × 10−6 cells/l, respectively). Anemia was noted in four patients (P2, P6, P8, and P10; 7.9, 10.9, 12.1, and 9.9 g/dl, respectively).
Decreased immunoglobulins (Ig) G levels were noted in four patients (P4, P5, P7, and P10; 315, < 30, 486, and 331 mg/dl; age-adjusted normal range 639–1439, 463–1236, 608–1572, and 700–1600 mg/dl, respectively). Decreased IgA was found in five patients (P4, P5, P6 P7, and P10; 34, < 5, < 5, < 5, and 74; 70–312, 25–154, 33–202, 45–236, and 70–400 mg/dl, respectively). Three patients had decreased IgM levels (P4, P5, and P6; 15, < 5, and 43; 56–352, 43–196, and 48–207 mg/dl, respectively). Coombs test was positive in four patients (P1, P2, P5, and P10). Other autoantibodies were found positive in two patients and included anti-nuclear antibodies + 1 to + 2 out of four (P2) and anti-thyroperoxidase antibodies > 1000 IU/ml (P8; normal range < 35 IU/ml).
Genetic diagnosis of LRBA deficiency
WES revealed three LRBA gene mutations (Table 1). P1, P2, P3, and P6 were found to have the same mutation (c.8174_8175insCATG, p.N2727Hfs*13). P4, P5, P7, P8, and P9 were found to have another mutation (c.7970T>G, p.I2657S). P10 had a different mutation (c.7869_7873delTTCTA, p.S2624Rfs*23). Sanger sequencing confirmed mutations in all patients. The families reported in our study are not related, and genetic analysis results were attributed to a possible founder effect in the Palestinian population.
Pulmonary manifestations and workup
Pulmonary manifestations at initial presentation
Pulmonary manifestations at initial presentation are presented in Table 2. Four patients (P3, P6, P8, and P9) presented with no respiratory complaints. Chronic cough was noted in six patients on their first admission. One patient (P1) had dyspnea and perioral cyanosis at rest. One of the patients (P5) had clubbing. Lung auscultation on initial presentation was normal in six patients, with good bilateral aeration, and clear lung fields. Unilateral left-sided crackles were noted in two patients (left upper and left lower lobes, P2 and P5, respectively). Decreased bilateral air entry was noted in two other patients (P1 and P7). Examination of P1 revealed diffuse bilateral wheezing and crackles.
Pulmonary workup
Pulmonary workup was guided by the clinical presentation. Baseline PFT and CXR were available in five patients each. Bronchoscopy studies were available in four patients (Tables 2 and 3). CT scans were available in six patients (Table 2).
Chest X-rays and computed tomography scans
CXR were consistent with hilar lymphadenopathy in four patients (P1, P5, P7, and P10). Atelectasis was noted in one patient (P1). Consolidations were seen in two patients (P7 and P10).
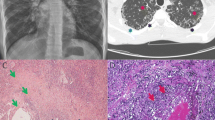
Representative CT scans are shown in Fig. 1. Opacifications suggestive of consolidations were seen in five (50%) patients. Lobar and sub-segmental atelectasis were noted in two patients (P5 and P1, respectively). Two patients demonstrated bronchiectatic changes (P5 and P7). Lymphadenopathy in thoracic CT scans was observed in five patients. Axillary, mediastinal, and hilar lymphadenopathy were demonstrated in two, three, and two patients, respectively (Fig. 1a). In two patients (P7 and P10), CT scan demonstrated changes consistent with diffuse interstitial lung disease (ILD) including septal thickening, heterogeneous attenuation pattern, and patchy opacities (Fig. 1b). Ground glass opacities (GGOs) were seen in three patients (P1, P2, and P7; Fig. 1e). Lung nodules were seen in five patients (P1, P2, P4, P7, and P10; Fig. 1f). In three of them (P1, P2, and P4), only two to five lung nodules were seen in each patient.

Computed tomography (CT) scans of LRBA-deficient patients. a, b Baseline axial images of chest CT of P7 in mediastinal (a) and lung (b) windows demonstrating severe lymphadenopathy and diffuse interstitial lung changes. c, d Corresponding axial images of P7 in mediastinal (c) and lung (d) windows following abatacept treatment with marked improvement of the findings shown in a, b. e Axial image of chest CT of P1 in lung window. Noted are ground glass opacities in the right upper lobe (arrow). f Axial image of chest CT of P1 in lung window. Sub-segmental atelectasis in the right middle lobe and small nodules at the periphery of both lower lobes are remarkable (arrows)
Pulmonary function testing
Presentation PFT included spirometry in five patients (Table 3). Lung volumes, airway resistance, and diffusion capacity were available in three patients (P1, P4, and P10). One patient had normal flows with normal high lung volumes and normal airway resistance (P4) but increased residual volume, likely secondary to poor effort. The remaining four (P1, P2, P7, and P10) had a symmetrical forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) decrease in flows. One of which had normal lung volumes and airway resistance. The corrected diffusion capacity was normal. All patients were young children with no prior experience with the test, and therefore, technical difficulties were common. One patient (P6) completed PFT on a follow-up visit. However, she managed less than a 1-s effort, and the test was therefore considered uninterpretable and is not displayed in the table. In summary, these pulmonary functions are grossly normal with no clear evidence of obstructive or restrictive disease.
Bronchoscopy studies
Patients with respiratory complaints and abnormal imaging as described above underwent fiber-optic bronchoscopy with BAL (Table 2). Normal respiratory tract anatomy was noted in two out of four patients. Tracheomalacia and adenoid hypertrophy were seen in two and one patients, respectively.
Pulmonary cytology and pathology
Cytological examination of the BAL fluid was available in all the patients that underwent bronchoscopy (P1, P2, P4, and P7). In all the patients above, specific pathogens were not identified. A few inflammatory cells (IC) and alveolar macrophages (AM) were seen in the BAL specimens of P2 and P4, respectively. The BAL of P7 demonstrated an abundant amount of IC and AM. In P7, a trans-bronchial biopsy was performed. Pathological examination revealed a dense chronic inflammation in the bronchial wall. In the lung parenchyma, chronic inflammation was noted in the alveolar septae with fibrin and neutrophils in the alveolar spaces. No granulomas were seen.
Microbiological analysis of the respiratory tract
Cultures from the respiratory tract were available in six patients (P1, P2, P4, P5, P7, and P10). BAL cultures were positive for Haemophilus influenzae beta-lactamase negative in two patients (P4 and P7).
Coughed sputum cultures were positive for Haemophilus influenzae beta-lactamase negative in three patients (P2, P5, and P10) and Streptococcus pneumoniae in one patient (P5). Sputum cultures in P2 were taken 7 months following BAL cultures. P5 and P10 did not undergo bronchoscopy and had no BAL cultures available. Galactomannan was available in two patients (P7 and P10) and was positive in one (P10).
Polymerase chain reaction (PCR) for respiratory viruses was available in three patients (P4, P7, and P10) and positive in one (P7 for influenza virus type A (H1N1), cytomegalovirus (CMV; 810 copies/ml) and herpes simplex virus-1. During hospitalization, in which bronchoscopy was performed, P7 continued to have chronic productive cough and thus treatment with amoxicillin and clavulanic acid, azithromycin and oseltamivir was started. P4 and P5 were treated with amoxicillin due to chronic cough and growth in cultures described above.
Treatment and outcome
Treatment and outcome are presented in Table 1. Treatment was directed against LRBA deficiency-related symptoms. Seven patients received pharmacotherapy directed against autoimmune phenomena or hypogammaglobulinemia. One patient (P8) received methimazole for Graves’ disease. Six patients (P1, P2, P4, P5, P7, and P10) received monthly courses of intravenous immunoglobulins (IVIG). Immunosuppressive drugs were given to five patients and included glucocorticosteroids (GCS; P1, P2, P4, P7, and P10), rapamycin (P1, P2, and P4), and mycophenolate mofetil (MMF; P1, P2, and P4). One patient (P7) was treated with abatacept and hydroxychloroquine. P10 previously received several courses of rituximab for autoimmune cytopenia.
Mean follow-up period (excluding P8 and P9, who have continued follow-up in another medical center) was 4.5 (range 0.4–14.4) years. Mean current age is 12.07 (range 4.6–25.5) years. In all patients, respiratory symptoms have appeared before receiving treatment and did not significantly change during the treatment. All patients are currently alive with a stable clinical status. In their last physical examination, cough was noted in three patients (P4, P5, and P10). Findings on physical examination were noted in three patients (left lung crackles and upper respiratory rhonchi, P5 and P7, respectively, and decreased RUL air entry and wheeze in P10).
Two patients (P4 and P7) underwent hematopoietic stem cell transplantation (HSCT), and both are currently alive and well.
Pulmonary symptoms of P7 improved following treatment with abatacept. A 2-month follow-up CT scan, which was done before her HSCT and on antibiotic and abatacept treatment, has demonstrated a significant reduction in mediastinal lymphadenopathy and interstitial changes (Fig. 1c, d, respectively). P10 is the oldest patient in our cohort. He suffers a chronic productive cough, with occasional hemoptysis with musculoskeletal chest pain. He has recently been commenced on inhaled salbutamol and fluticasone with an improvement in his symptoms, and is currently preparing for HSCT.
Discussion
In this study, we summarize respiratory features and workup of 10 patients with genetically proven LRBA deficiency.
LRBA deficiency was first reported in four families, who suffered from hypogammaglobulinemia and autoimmunity [19]. In a cohort of 22 patients, clinical characteristics consisted of immune dysregulation, hepatosplenomegaly, recurrent infections, hypogammaglobulinemia, and reduced counts of regulatory T (Treg) and B cells [13]. Other studies have extended the clinical phenotype of LRBA deficiency [2, 3, 5, 22]. However, studies focusing on respiratory manifestations in these patients are lacking.
LRBA deficiency is a rare autosomal recessive disease, and data regarding epidemiology is extracted from case series and small cohorts. High prevalence is noted among patients from communities with high rate of consanguineous marriages. However, compound heterozygous cases have also been described [13].
In our study, pneumonia, as confirmed by CT scans and isolation of bacterial pathogens from the respiratory tract, was noted in 50% of the patients. Combined immunodeficiency is characteristic of LRBA deficiency and explains this observation [1]. Prevalence of respiratory tract infections is reported in a review of 31 LRBA-deficient patients to be 61% [2]. Other studies reported an even higher incidence, such as 76 and 80% among 17 and 22 LRBA-deficient patients, respectively [6, 13].
Bronchiectasis was found in 20% of our patients. This corresponds well with previous studies, which reported bronchiectasis in one third of LRBA-deficient patients [2]. As both patients with bronchiectasis in our study had a history of recurrent pneumonia, bronchiectasis is probably a manifestation of post-infectious sequela. PID is gaining appreciation in the aetiopathogenesis of childhood non-cystic fibrosis bronchiectasis, with an incidence rate that varies between 9.1 and 17% [7]. Changes in diversity and composition of lung microbiome, partly resulting from recurrent pulmonary infections and chronic inflammatory processes, may play a key role in bronchiectasis formation [9]. Interestingly, one of our patients presented with digital clubbing, a feature already described in LRBA-deficient patients, with an incidence rate of 26% in one study [2] and 65% in another [6].
Non-infectious, immune-mediated pulmonary manifestations were found in several of our patients. This includes ILD, GGOs, and lung nodules. Of the 22 LRBA-deficient patients reported in one study, 38% presented with granulomatous-lymphocytic ILD (GLILD) [13]. Lower rates of 6 and 18% were noted in other studies [2, 6]. GLILD was previously reported in common variable immunodeficiency disorder (CVID) [17, 21, 26], with immune complexes and switched memory B cells suggested to be involved in pathogenesis [21].
LRBA deficiency belongs to PID characterized by regulatory T cells (Treg) dysfunction. This group includes diseases such as immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, cytotoxic T lymphocyte antigen-4 (CTLA-4) deficiency, and signal transducer and activator of transcription (STAT)1 gain of function (GOF) [28]. In these conditions, pulmonary disease occurs not only secondary to infection, but also due to the autoimmune sequelae of Treg dysfunction (ILD, granuloma, fibrosis). For example, GLILD was reported in patients with CTLA-4 deficiency [24], bronchiectasis in STAT1 GOF mutation [10], and acute respiratory distress syndrome in IPEX syndrome [8]. In such conditions, pulmonary manifestations are likely to be improved by general measures such as IVIG replacement, anti-microbial and immunosuppressive therapy, and also targeted therapy if it exists for the specific deficiency, as in the case of abatacept for LRBA and CTLA4 deficiency [18].
Another key finding in our study is thoracic lymphadenopathy, as evident in CT scans of 50% of our patients. Autoimmune lymphoproliferative syndrome-like presentation of LRBA deficiency was previously reported in four of our patients [13, 22].
The pulmonary functions the patients performed did not reveal an obstructive or restrictive disease and thus show no evidence of airway or interstitial lung disease. This is consistent with the finding of mostly infectious pulmonary diseases. However, interpretation was compounded by poor technique and effort in the majority of patients.
An important retrospective multicenter study examined the pulmonary monitoring of 178 pediatric PID patients receiving IVIG treatment and found a significant variation between different European centers. Lack of international guidelines and standardization of biomarkers for disease progression were noted as possible reasons [15]. Pulmonary workup and monitoring in our 10 LRBA patients were clinically guided, as no such consensus guidelines are available.
Of the 10 patients in our study, 6 required treatment and the remaining are under surveillance alone. Treatment was largely composed of immunosuppression. GCS and rapamycin achieved and maintained clinical remission in most cases. MMF was used to taper down GCS treatment in three patients with partial clinical response. Treatment of one patient with abatacept, a CTLA4-Ig fusion agent, was found to be effective in ameliorating pulmonary symptoms and findings in CT, as previously published [18].
Our experience with HSCT in LRBA deficiency is limited with two patients. Prior studies reported successful treatment with allogeneic HSCT with an overall survival rate of 67% [25]. As reported, autoimmune phenomena in LRBA deficiency have been shown to improve following HSCT [23, 27].
Our study has few limitations. It is small sized and retrospective, although all reported cohorts of LRBA-deficient patients are small [5, 13, 22]. The largest cohort summarized so far to the best of our knowledge consisted of 31 patients [2]. Despite these limitations, we have demonstrated that even young children with LRBA deficiency often suffer respiratory symptoms and pulmonary changes. The oldest patient in our study, at 25 years old, demonstrates the potential prognosis of essentially untreated long-term LRBA lung disease: chronic productive cough, bullous changes, lung nodules, ILD, and pulmonary lymphadenopathy. Therefore, based on the clinical presentation and high rate of pulmonary complications in these patients, we would suggest a baseline CT scan for all newly diagnosed patients.
In conclusion, clinical characteristics of LRBA deficiency often include different pulmonary manifestations, such as respiratory tract infections, bronchiectasis, and ILD, which must be investigated, monitored, and treated from a young age. Awareness of pulmonary morbidity in these patients and involvement of a pulmonologist in the workup and clinical decision-making are keys to effective treatment.
Abbreviations
- AM:
-
Alveolar macrophages
- BAL:
-
Bronchoalveolar lavage
- CMV:
-
Cytomegalovirus
- CT:
-
Computed tomography
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen-4
- CVID:
-
Common variable immunodeficiency disorder
- CXR:
-
Chest X-rays
- FEV1:
-
Forced expiratory volume in 1 s
- FTT:
-
Failure to thrive
- FVC:
-
Forced vital capacity
- GCS:
-
Glucocorticosteroids
- GGOs:
-
Ground glass opacities
- GLILD:
-
Granulomatous-lymphocytic ILD
- HSCT:
-
Hematopoietic stem cell transplantation
- IC:
-
Inflammatory cells
- Ig:
-
Immunoglobulins
- ILD:
-
Interstitial lung disease
- IPEX:
-
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked
- IVIG:
-
Intravenous Ig
- LPS:
-
Lipopolysaccharide
- LRBA:
-
LPS-responsive beige-like anchor
- MMF:
-
Mycophenolate mofetil
- PFT:
-
Pulmonary function testing
- Treg:
-
Regulatory T cells
- WES:
-
Whole exome sequencing
References
Al Sukaiti N, AbdelRahman K, AlShekaili J, Al Oraimi S, Al Sinani A, Al Rahbi N, Cho V, Field M, Cook MC (2017) Agammaglobulinaemia despite terminal B-cell differentiation in a patient with a novel LRBA mutation. Clin Transl Immunology 6:e144
Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, Mohammadzadeh I, El-Rajab MA, Massaad M, Chou J, Aghamohammadi A, Geha RS, Hammarström L (2016) Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol 36:33–45
Al-Mayouf SM, Naji H, Alismail K, Alazami AM, Sheikh F, Conca W, Al-Mousa H (2017) Evolving spectrum of LRBA deficiency-associated chronic arthritis: is there a causative role in juvenile idiopathic arthritis? Clin Exp Rheumatol 35:327–329
Azizi G, Abolhassani H, Mahdaviani SA, Chavoshzadeh Z, Eshghi P, Yazdani R, Kiaee F, Shaghaghi M, Mohammadi J, Rezaei N, Hammarström L, Aghamohammadi A (2017) Clinical, immunological, molecular analyses and outcomes of Iranian patients with LRBA deficiency: a longitudinal study. Pediatr Allergy Immunol 28:478–484
Azizi G, Abolhassani H, Mahdaviani SA, Chavoshzadeh Z, Eshghi P, Yazdani R, Kiaee F, Shaghaghi M, Mohammadi J, Rezaei N, Hammarström L, Aghamohammadi A (2017) Clinical, immunologic, molecular analyses and outcomes of Iranian patients with LRBA deficiency: a longitudinal study. Pediatr Allergy Immunol 28:478–484
Azizi G, Abolhassani H, Mahdaviani SA, Chavoshzadeh Z, Eshghi P, Yazdani R, Kiaee F, Shaghaghi M, Mohammadi J, Rezaei N, Hammarstrom L, Aghamohammadi A (2017) Clinical, immunologic, molecular analyses and outcomes of iranian patients with LRBA deficiency: a longitudinal study. Pediatr Allergy Immunol 28:478–484
Bahçeci S, Karaman S, Nacaroğlu HT, Yazıcı S, Girit S, Ünsal-Karkıner Ş, Can D (2016) Changing epidemiology of non-cystic fibrosis bronchiectasis. Turk J Pediatr 58:19–26
Baris S, Schulze I, Ozen A, Karakoc Aydiner E, Altuncu E, Karasu GT, Ozturk N, Lorenz M, Schwarz K, Vraetz T, Ehl S, Barlan IB (2014) Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X-linked: pulmonary involvement as a non-classical disease manifestation. J Clin Immunol 34:601–606
Boyton RJ, Reynolds CJ, Quigley KJ, Altmann DM (2013) Immune mechanisms and the impact of the disrupted lung microbiome in chronic bacterial lung infection and bronchiectasis. Clin Exp Immunol 171:117–123
Breuer O, Daum H, Cohen-Cymberknoh M, Unger S, Shoseyov D, Stepensky P, Keller B, Warnatz K, Kerem E (2017) Autosomal dominant gain of function STAT1 mutation and severe bronchiectasis. Respir Med 126:39–45
Burnett DL, Parish IA, Masle-Farquhar E, Brink R, Goodnow CC (2017) Murine LRBA deficiency causes CTLA-4 deficiency in Tregs without progression to immune dysregulation. Immunol Cell Biol 95:775–788
Charbonnier LM, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, Massaad MJ, Garcia-Lloret M, Hanna-Wakim R, Dbaibo G, Alangari AA, Alsultan A, Al-Zahrani D, Geha RS, Chatila TA (2015) Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol 135:217–227
Gamez-Diaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, Morio T, Worth AJ, Blessing J, Van de Veerdonk F, Feuchtinger T, Kanariou M, Schmitt-Graeff A, Jung S, Seneviratne S, Burns S, Belohradsky BH, Rezaei N, Bakhtiar S, Speckmann C, Jordan M, Grimbacher B (2016) The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol 137:223–230
Hou TZ, Verma N, Wanders J, Kennedy A, Soskic B, Janman D, Halliday N, Rowshanravan B, Worth A, Qasim W, Baxendale H, Stauss H, Seneviratne S, Neth O, Olbrich P, Hambleton S, Arkwright PD, Burns SO, Walker LS, Sansom DM (2017) Identifying functional defects in patients with immune dysregulation due to LRBA and CTLA-4 mutations. Blood 129:1458–1468
Jolles S, Sánchez-Ramón S, Quinti I, Soler-Palacín P, Agostini C, Florkin B, Couderc LJ, Brodszki N, Jones A, Longhurst H, Warnatz K, Haerynck F, Matucci A, de Vries E (2017) Screening protocols to monitor respiratory status in primary immunodeficiency disease: findings from a European survey and subclinical infection working group. Clin Exp Immunol 190:226–234
Levy E, Stolzenberg MC, Bruneau J, Breton S, Neven B, Sauvion S, Zarhrate M, Nitschke P, Fischer A, Magerus-Chatinet A, Quartier P, Rieux-Laucat F (2016) LRBA deficiency with autoimmunity and early onset chronic erosive polyarthritis. Clin Immunol 168:88–93
Lieberman P, Routes J (2014) Granulomatous-lymphocytic interstitial lung disease in a patient with common variable immunodeficiency. J Allergy Clin Immunol Pract 2:824
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, Fritz JM, Marsh R, Husami A, Kissell D, Nortman S, Chaturvedi V, Haines H, Young LR, Mo J, Filipovich AH, Bleesing JJ, Mustillo P, Stephens M, Rueda CM, Chougnet CA, Hoebe K, McElwee J, Hughes JD, Karakoc-Aydiner E, Matthews HF, Price S, Su HC, Rao VK, Lenardo MJ, Jordan MB (2015) AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349:436–440
Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, Srugo I, Melamed D, Hultenby K, Liu C, Baronio M, Vitali M, Philippet P, Dideberg V, Aghamohammadi A, Rezaei N, Enright V, du L, Salzer U, Eibel H, Pfeifer D, Veelken H, Stauss H, Lougaris V, Plebani A, Gertz EM, Schäffer AA, Hammarström L, Grimbacher B (2012) Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 90:986–1001
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J, Force AET (2005) Standardisation of spirometry. Eur Respir J 26:319–338
Park JH, Levinson AI (2010) Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol 134:97–103
Revel-Vilk S, Fischer U, Keller B, Nabhani S, Gámez-Díaz L, Rensing-Ehl A, Gombert M, Hönscheid A, Saleh H, Shaag A, Borkhardt A, Grimbacher B, Warnatz K, Elpeleg O, Stepensky P (2015) Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation. Clin Immunol 159:84–92
Sari S, Dogu F, Hwa V, Haskologlu S, Dauber A, Rosenfeld R, Polat M, Kuloglu Z, Kansu A, Dalgic B, Ikinciogullari A (2016) A successful HSCT in a girl with novel LRBA mutation with refractory celiac disease. J Clin Immunol 36:8–11
Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schäffer AA, Grüning BA, Unger S, Frede N, Baumann U, Witte T, Schmidt RE, Dueckers G, Niehues T, Seneviratne S, Kanariou M, Speckmann C, Ehl S, Rensing-Ehl A, Warnatz K, Rakhmanov M, Thimme R, Hasselblatt P, Emmerich F, Cathomen T, Backofen R, Fisch P, Seidl M, May A, Schmitt-Graeff A, Ikemizu S, Salzer U, Franke A, Sakaguchi S, Walker LSK, Sansom DM, Grimbacher B (2014) Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 20:1410–1416
Seidel MG, Bohm K, Dogu F, Worth A, Thrasher A, Florkin B, Ikinciogullari A, Peters A, Bakhtiar S, Meeths M, Stepensky P, Meyts I, Sharapova SO, Gamez-Diaz L, Hammarstrom L, Ehl S, Grimbacher B, Gennery AR (2017) Treatment of severe forms of LPS-responsive beige-like anchor protein deficiency with allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol 141:770–775
Shah JL, Amin SB, Verma N, Mohammed TL (2017) Granulomatous-lymphocytic interstitial lung disease in a patient with common variable immunodeficiency. Curr Probl Diagn Radiol
Tesi B, Priftakis P, Lindgren F, Chiang SC, Kartalis N, Löfstedt A, Lörinc E, Henter JI, Winiarski J, Bryceson YT, Meeths M (2016) Successful hematopoietic stem cell transplantation in a patient with LPS-responsive beige-like anchor (LRBA) gene mutation. J Clin Immunol 36:480–489
Verbsky JW, Chatila TA (2013) Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr Opin Pediatr 25:708–714
Acknowledgments
We would like to thank our departmental nursing and administrative staff for their tireless commitment to patient care. We would also like to thank Prof. Zeev Rotstein, Director of The Hadassah Medical Center, for his support of the department and the patients.
Funding
Bella Shadur’s position is supported by the Australian Government Research Training Program Scholarship. This work was supported by the Deutsche Forschungsgemeinschaft (Discovery and Evaluation of New Combined Immunodeficiency Disease Entities; grant DFG WA 1597/4-2) and the ERA-Net ERARE consortium EURO-CID.
Author information
Authors and Affiliations
Contributions
All authors have read and approved this manuscript. Oded Shamriz* gathered the data and wrote of manuscript, Bella Shadur*, Adeeb NaserEddin, and Irina Zaidman treated the patients and gathered data, Natalia Simanovsky gave radiological consultation, Orly Elpeleg supervised genetic analysis, Eitan Kerem and Joel Reiter gave pulmonologist consultation, Polina Stepensky treated the patients, designed, and supervised the study.
Corresponding author
Ethics declarations
The study was approved by the Hadassah and Israeli Ministry of Health institutional review boards (number 187-17-HMO).
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
Not applicable.
Clinical trials registration
Not applicable.
Additional information
Communicated by Piet Leroy
Rights and permissions
About this article

Cite this article
Shamriz, O., Shadur, B., NaserEddin, A. et al. Respiratory manifestations in LPS-responsive beige-like anchor (LRBA) protein-deficient patients. Eur J Pediatr 177, 1163–1172 (2018). https://doi.org/10.1007/s00431-018-3171-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-018-3171-5